

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website https://www.gastrores.org |
Review
Volume 15, Number 1, February 2022, pages 1-12

Statin Use in Patients With Chronic Liver Disease and Cirrhosis: Current Evidence and Future Directions
Malek Kreidieha, Rachelle Hamadia, Mira Alsheikhb, Hassan Al Moussawib, Liliane Deebb, c
aDepartment of Internal Medicine, Staten Island University Hospital, Northwell Health, Staten Island, NY, USA
bDivision of Gastroenterology, Department of Internal Medicine, Staten Island University Hospital, Northwell Health, Staten Island, NY, USA
cCorresponding Author: Liliane Deeb, Division of Gastroenterology, Department of Internal Medicine, Staten Island University Hospital, Northwell Health, Staten Island, NY, USA
Manuscript submitted December 28, 2021, accepted January 5, 2022, published online February 25, 2022
Short title: Statin Use in Cirrhosis
doi: https://doi.org/10.14740/gr1498
- Abstract
- Introduction
- Search for New Therapeutic Strategies to Prevent Cirrhosis Development and Its Decompensation
- Statins
- Main Pathways Regulated by Statins in Liver Cells
- Preclinical Data on the Use of Statins in Animal Models
- Clinical Data on the Use of Statins in Patients With CLD
- Conclusions and Future Directions
- References
| Abstract | ▴Top |
Chronic liver disease (CLD) and its complications constitute a significant cause of mortality and morbidity worldwide. Most deaths are secondary to the decompensation of cirrhosis and evolution of portal hypertension (PHTN). Since disease progression reversal is hardly attainable after decompensated cirrhosis develops, it is essential to intervene early with a therapeutic agent or regimen that could prevent or slow disease evolution. Thus far, there has been no agreed-upon medication to help in the fight against the development of cirrhosis or its decompensation. While early data depicted statins as harmful agents for the liver, current evidence from preclinical and clinical studies suggests that they might have positive impact on CLD. Low-quality evidence supports the fact that statins reduce mortality in CLD. Moderate-quality evidence suggests that statins reduce the risk of hepatic decompensation, variceal bleeding, and mortality, especially among patients with compensated cirrhosis. Combining this data with the long track-record of safety and tolerability of statins and their potential benefits in hepatocellular carcinoma (HCC) risk reduction, hepatologists might soon rely on statins to achieve better outcomes in their CLD and cirrhotic patients without significant additional costs. This review describes the rationale behind the use of statins in patients with CLD and cirrhosis. It sheds light on the current preclinical and clinical studies that reflect beneficial effects of the use of different types and doses of statins in the treatment of patients with different types and stages of CLD and cirrhosis. It also emphasizes the need for designing and developing additional large prospective interventional randomized control trials (RCTs) to better evaluate the association between statin exposure and the risk of fibrosis progression and development of cirrhosis in patients with non-cirrhotic CLDs, the risk of progression of PHTN in patients with cirrhosis, and the mortality rates in patients with cirrhotic or non-cirrhotic CLDs.
Keywords: Statins; Chronic liver disease; Cirrhosis; Portal hypertension; Hepatocellular carcinoma
| Introduction | ▴Top |
Chronic liver disease (CLD) and its complications constitute a significant cause of mortality and morbidity worldwide. According to the Center for Disease Control, they continue to rank among the top 15 causes of mortality in the USA [1]. According to the Global Burden of Disease study, 1.32 million patients passed away due to cirrhosis-related causes globally in 2017, and this constituted around 2.4% of global deaths [2]. Most deaths in cirrhosis are secondary to the decompensation of cirrhosis and evolution of portal hypertension (PHTN).
PHTN occurs in around 90% of cirrhotic patients [3], and its evolution in patients with decompensated cirrhosis results in complications such as ascites, hepatorenal syndrome (HRS), hepatopulmonary syndrome (HPS), spontaneous bacterial peritonitis (SBP), splenomegaly, gastroesophageal varices, and hepatocellular carcinoma (HCC) [4-6]. Among these, gastroesophageal varices are responsible for almost 25% of deaths in cirrhosis due to their tendency to rupture and bleed [7, 8].
Since disease progression reversal is hardly attainable after decompensated cirrhosis develops, it is essential to intervene early with a therapeutic agent or regimen that could prevent or slow the disease evolution. Thus far, specific therapies against hepatitis B and C, autoimmune hepatitis, and other etiologies of cirrhosis have been used in the fight against the development of cirrhosis or its decompensation.
| Search for New Therapeutic Strategies to Prevent Cirrhosis Development and Its Decompensation | ▴Top |
To better understand the pathophysiology behind the development of PHTN in cirrhotic patients, several advances have been recently made. While it was initially speculated that PHTN results mainly from the distortion in anatomy and function of normal hepatic tissue [9, 10], it is currently established that the amelioration of portal inflow that results from splanchnic vasodilation also tends to have a significant impact on its development [9, 11, 12].
As a result of these advances in the understanding of the pathophysiology of PHTN, a myriad of therapeutic agents, including non-selective beta-blockers and carvedilol, haven been developed and have proven to have positive prognostic implications in cirrhotic patients. This opened the way for epidemiological studies to test hypothetical benefits of new therapeutic strategies, such as the use of statins, in targeting different stages along the transition from CLD to cirrhosis on one hand, and the transition from compensated cirrhosis to decompensated cirrhosis on the other.
| Statins | ▴Top |
Interestingly, statins, which are well known for their cardioprotective effects [13-16], are currently being evaluated for their effects on cirrhosis progression. These lipid-lowering agents, which act through the competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, are usually under-prescribed in cirrhotic patients due to concern of hepatotoxicity [17]. This has been reflected by the under-prescription of statins in CLD patients who have clear indications for statins, such as dyslipidemia or cardiovascular disease.
In recent years, several epidemiological studies have shown that statins have benefits other than those related to primary or secondary prevention of atherosclerotic disease. These are referred to as the pleiotropic effects and have been reflected in conditions such as CLD, chronic obstructive pulmonary disease, acute kidney injury, contrast-induced nephropathy, pancreatitis, erectile dysfunction, and others [18]. When it comes to CLD, data have shown that a reduction in portal pressure, liver sinusoidal endothelial cell (LSEC) and hepatic microvascular dysfunction, fibrogenesis, and sensitivity to endotoxin-mediated liver damage might play a role in preventing cirrhosis and delaying its progression [6, 19-21]. It was speculated that statins can contribute to this via a reduction in oxidative stress with a resultant decrease in the activation of inflammatory cells [22-26]. This has also been thought to be related to the activation of nitric oxide (NO) synthesis with a resultant improvement in endothelial function and number of endothelial progenitor cells [27-32].
In the light of the recent evidence regarding the unexpectedly beneficial effects of statins in cirrhosis, their use in cirrhotic patients has evolved from being risky to possibly promising [33]. As a result, many studies have been designed to further characterize such theoretically favorable effects in the primary and secondary prevention of cirrhosis [34, 35]. In the following review article, we will summarize current evidence regarding the influence of statins on fibrosis progression and cirrhosis development and evolution.
| Main Pathways Regulated by Statins in Liver Cells | ▴Top |
Rac1/KLF2 pathway
The transcription factor Kruppel-like factor 2 (KLF2) is a key component of the hepatic endothelium regulating around 40% of the endothelial genome and several processes such as inflammation, fibrosis, apoptosis, oxidative stress, vasodilation, and thrombosis [36, 37]. Within the liver, the expression of KLF2 occurs in major cell types including LSECs, hepatic stellate cells (HSCs), hepatocytes, and macrophages.
In general, statins upregulate KLF2 expression through the Rac1-MEK5-ERK5-MEF2 pathway resulting in HSC deactivation and inhibition of migration and proliferation (Figs. 1, 2) [38, 39]. Within the liver, the activation of the KLF2 pathway by statins has been demonstrated in animal models of hepatic injury [40].
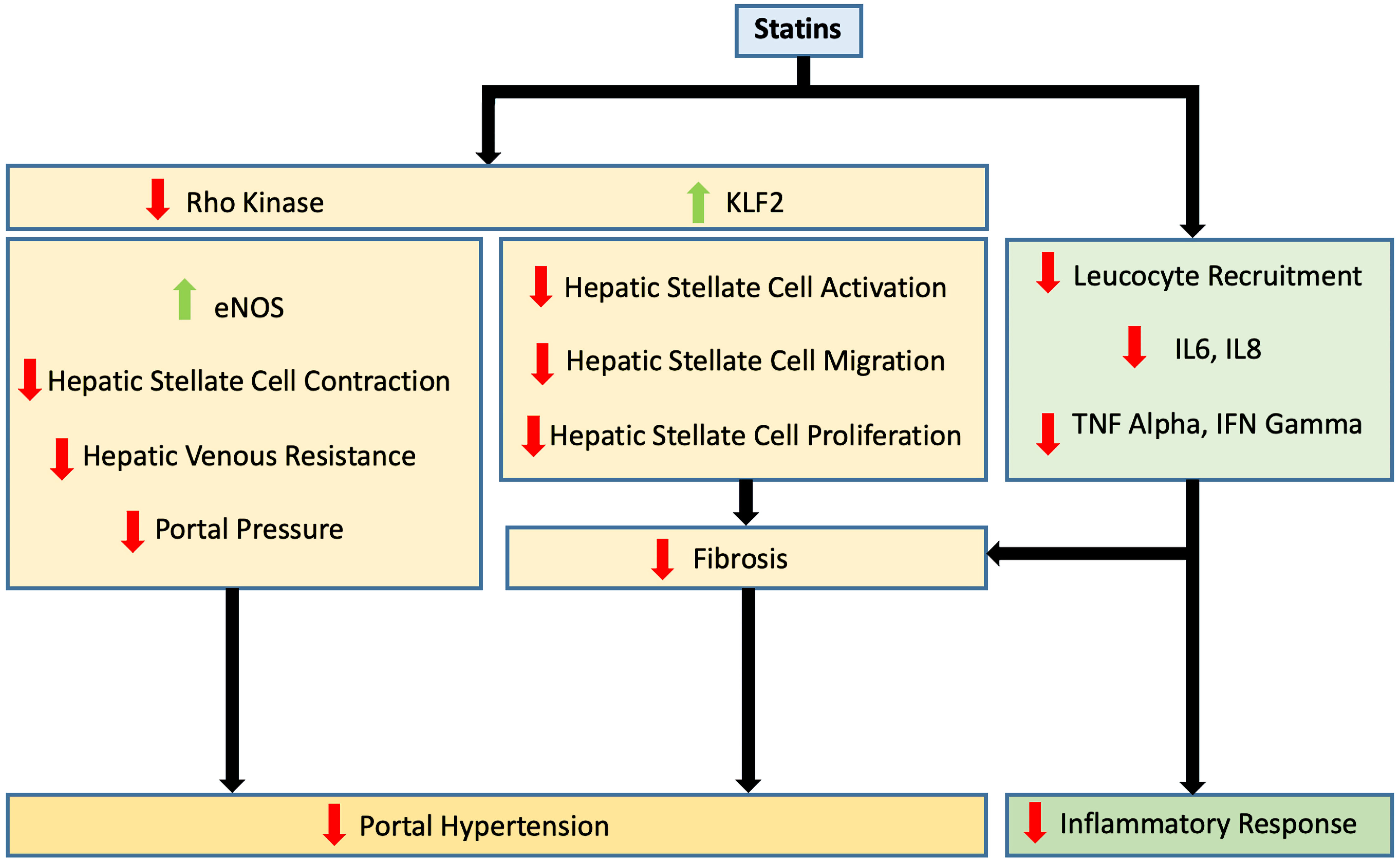 Click for large image | Figure 1. Effects of statin use on portal hypertension and inflammatory response. eNOS: endothelial nitric oxide synthase; IL: interleukin; IFN: interferon; KLF2: Kruppel-like factor 2; TNF: tumor necrosis factor. |
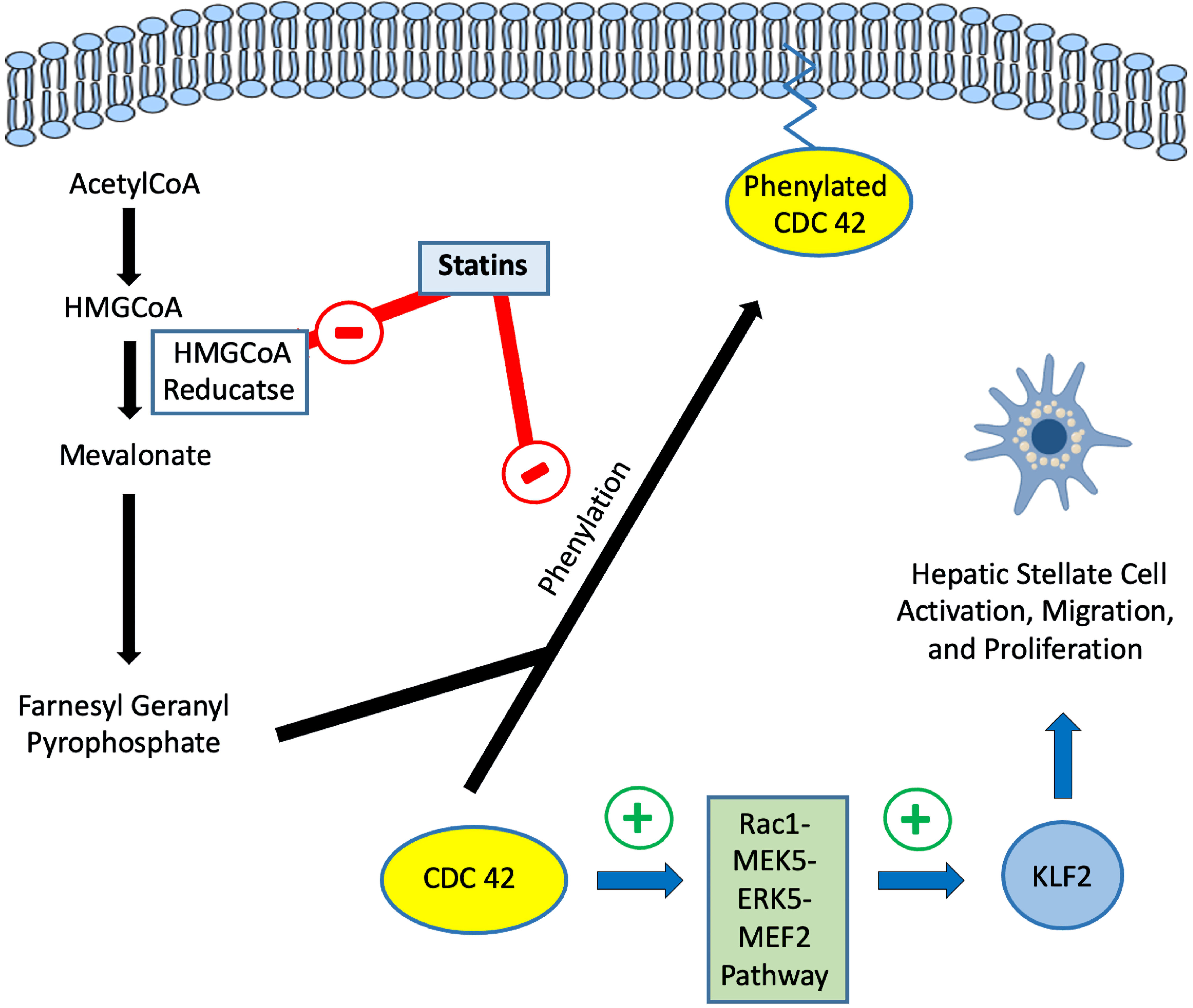 Click for large image | Figure 2. Regulation of Rac1/KLF2 pathway by statins in hepatic stellate cells. HMG-CoA: 3-hydroxy-3-methylglutaryl coenzyme A; KLF2: Kruppel-like factor 2. |
RhoA/Rho-kinase pathway
While statins improve endothelial functionality through the upregulation of KLF2, they ameliorate liver vascular pathobiology via the inhibition of Ras homolog gene family member A (RhoA)/Rho-kinase pathway. This has been demonstrated in animal models of cirrhosis [41]. By inhibiting RhoA/Rho-kinase signaling, statins inhibit the translocation of RhoA to its active site on the cell membrane, which prevents Rho-kinase from being fully activated. This in turn inhibits HSC contraction through the activation of myosin light chain phosphatase and the reduction in phosphorylated myosin. It also results in increased endothelial NO synthase (eNOS) activity and reduced hepatic venous resistance and portal pressure (Figs. 1, 3).
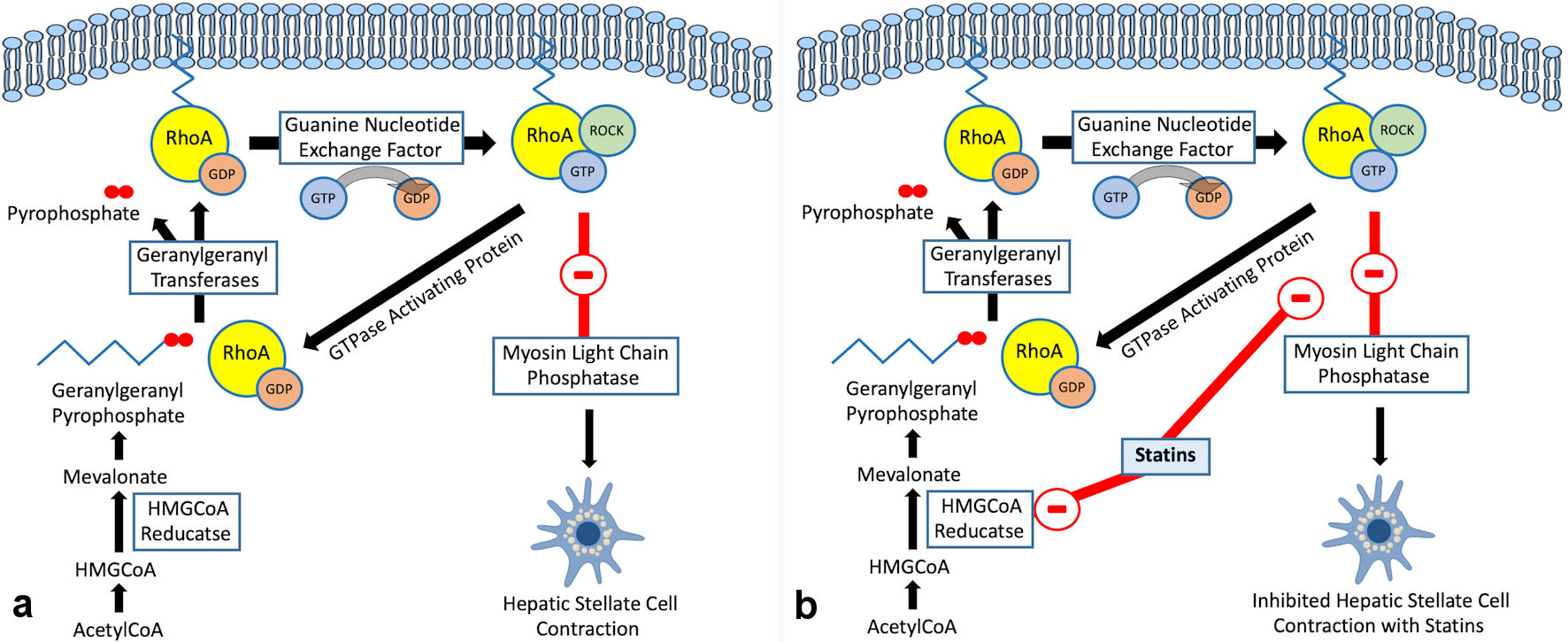 Click for large image | Figure 3. (a) RhoA/Rho-kinase pathway in hepatic stellate cells. (b) Regulation of RhoA/Rho-kinase pathway by statins in hepatic stellate cells. HMG-CoA: 3-hydroxy-3-methylglutaryl coenzyme A; RhoA: Ras homolog gene family member A. |
| Preclinical Data on the Use of Statins in Animal Models | ▴Top |
Preclinical data from studies performed on in vitro and in vivo animal models shed light on favorable effects of statins on endothelial function, angiogenesis, PHTN, and fibrosis. Suggested mechanisms involve regulation of KLF2, eNOS, thrombomodulin, and C-natriuretic peptide.
Liver fibrosis
Preclinical data support the fact that statins reduce liver fibrosis. For instance, early evidence suggests that the administration of fluvastatin, lovastatin, and simvastatin to primary rat hepatocytes and human hepatoma cell line inhibits the paracrine activation of HSCs [42]. Similarly, the administration of simvastatin to explanted rat livers has been shown to reduce oxidative stress, prevent liver damage, and improve endothelial dysfunction [43]. In addition, results from a preclinical study have demonstrated that the addition of atorvastatin to primary rat HSCs in vitro inhibits their differentiation to myofibroblasts and decreases collagen production and myofibroblast proliferation, thereby resulting in reduced turnover and fibrosis [44]. Another preclinical experiment by Trebicka et al revealed that while the early addition of atorvastatin to a bile duct ligation (BDL) cirrhosis animal model inhibits HSCs activation and fibrosis, its late use reduces HSC turnover and activity [45]. Interestingly, preclinical data on the complementary use of atorvastatin and losartan or pitavastatin and candesartan in cirrhotic rat models reflect an additive effect of such combinations on the inhibition and attenuation of liver fibrosis [46, 47].
In contrast, another study performed by Shirin et al on a rat model with thioacetamide-induced liver injury showed that neither atorvastatin nor rosuvastatin has an influence on HSC proliferation, liver fibrosis assessed by hydroxyproline content, or oxidative stress assessed by hepatic malondialdehyde levels [48].
PHTN
Preclinical data also support the fact that statins attenuate the increased hepatic vascular resistance by upregulating hepatic endothelial KLF2 and NO levels [40].
The role of KLF2 in PHTN has been highlighted through several preclinical studies on cirrhotic rat models. For instance, results from studies evaluating the addition of simvastatin to LSECs has demonstrated an upregulation in KLF2 expression with subsequent vasoprotective effects on hepatic vasculature [36, 39].
Similarly, the role of NO was recently highlighted when a NO-donating statin (NCX 6560) culminated in the lowering of portal pressure, with high vasoprotective properties and low toxicity rates in hepatocytes and myocytes [49]. In a study by Abraldes et al, carbon tetrachloride (CCl4)-induced cirrhotic rats treated with simvastatin had a selective increase in hepatic NO production, with a resultant improvement in LSEC dysfunction and a reduction in vasoconstriction [50]. Simvastatin has been also shown to induce NO-mediated vascular hypo-responsiveness in the setting of portal-systemic collateral vascular resistance and endothelin-1-related perfusion changes [51]. Evidence from studies evaluating the use of simvastatin in non-cirrhotic portal hypertensive PHTN animal models has been compatible with that obtained from studies evaluating its use in cirrhotic ones. In fact, simvastatin use in non-cirrhotic PHTN animal models has also been shown to upregulate eNOS, cyclooxygenase-2, and thromboxane A2 expression [52]. When it comes to atorvastatin, however, its use in cirrhotic and non-cirrhotic PHTN animal models has shown opposing effects. On the one hand, the use of atorvastatin in cirrhotic PHTN animal models has been shown to reduce the portal pressure, shunt flow, and angiogenesis by blocking the non-canonical Hedgehog (Hh) pathway, RhoA, in activated HSCs [41]. On the other hand, the use of atorvastatin in non-cirrhotic PHTN animal models has been proven to exacerbate non-cirrhotic PHTN and angiogenesis by augmenting the above-mentioned pathway [53, 54]. In contrast, when it comes to pravastatin, data from similar animal models with partial portal vein ligations have shown no effect on either the vascular response to endothelin-1 or on the systemic or portal hemodynamics [55].
Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)
In the light of NAFLD being an independent risk factor for metabolic syndrome and dyslipidemia, several preclinical studies have been developed to assess the effects of using statins on NAFLD progression and NASH development [56].
The use of statins in methionine-choline-deficient diet (MCDD)-fed mice has been shown to prevent the development of NASH by restoring the mitochondrial and peroxisomal fatty acid oxidation and by preventing the decreased expression of peroxisomal proliferator-activated receptor α (PPARα) induced by NASH [57].
When it comes to simvastatin, its administration into a NASH mouse model has led to the deactivation of the Ras and RhoA pathway with a subsequent reduction in hepatic inflammation and no significant changes in the intrahepatic cholesterol content or degree of steatosis [58]. Conflicting evidence was obtained when the use of simvastatin was studied in apolipoprotein E (ApoE) -/- mice which exhibit high tendency to develop atherosclerosis even on low-cholesterol diets due to reduced serum ApoE. For instance, results from simvastatin use in these ApoE mice have demonstrated an exacerbation in hepatic inflammation, steatosis, and fibrosis. The fact that they have described a resultant increase in oxidative stress under conditions of inflammatory stress suggests that inflammatory conditions in the liver might be a predisposing factor for statin-induced injury [59].
When it comes to rosuvastatin, its use in rats with NASH secondary to high-fat high-cholesterol diet has been shown to reduce histologic inflammation. This was postulated to be secondary to a reduction in the free fatty acid liver content and levels of inflammatory markers, such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, and alanine transaminase (ALT) [60].
Similarly, studies evaluating the effect of the novel statin subclass, pitavastatin, on NASH progression in estrogen-deficient ovariectomized mice with more severe histological lesions have resulted in a reduction in hepatic inflammation. The protective effect of pitavastatin was attributed to its down regulation of hepatic inflammatory genes, including c-c motif chemokine receptor 2 (CCR2), dichloromethylene (CCL2), TNF-α, and interferon (IFN)-γ [61].
| Clinical Data on the Use of Statins in Patients With CLD | ▴Top |
After the initial evidence from preclinical studies on the promising effects of statin use in CLD, an increasing number of epidemiological cross-sectional, retrospective, and prospective studies have been designed to evaluate the effects of statins on PHTN and cirrhosis development, progression, and decompensation. Unfortunately, however, some discrepancy in the data exists, and the strength of the evidence supporting the use of statins in CLD continues to be limited.
Retrospective studies commenting on the outcomes with the use of statins in cirrhosis
Increasing evidence that focuses on the mortality and morbidity benefit from statin use in cirrhosis has been accumulated [62, 63]. In one retrospective cohort study performed by Kumar et al with a mean follow-up of 36 months, the use of statins in 81 cirrhotic patients, who were mostly at Child-Pugh A stage and who were matched 1:2 with statin non-users, was associated with around 47% reduction in mortality and 37% reduction in hepatic decompensation [64]. It was, however, significantly associated with higher rates of NASH, diabetes mellitus, and coronary artery disease. Similarly, another retrospective cohort study performed using the Veteran Affairs Clinical Case Registry showed that the use of statins in 685 patients with hepatitis C virus (HCV)-related compensated cirrhosis, who were matched 1:5 with statin non-users, was associated with around 44% reduction in mortality and 45% reduction in decompensation episodes. After adjusting for FIB-4 index score, age, model for end-stage liver disease (MELD) score, serum level of albumin, and Child-Turcotte-Pugh score, the beneficial effects of statins on cirrhosis decompensation and mortality persisted at 10 years [65].
When it comes to the effect of statins on the incidence of infections requiring hospitalizations, a retrospective study was performed to compare rates of hospitalizations secondary to serious infections among veteran patients with compensated cirrhosis who were on statins on one hand or no statins on the other. After a mean follow-up of 1,194 days, the 154 patients who were on statins had a hazard ratio (HR) of 0.67 for serious infections requiring hospitalizations [66].
In order to better understand the morbidity and mortality benefits of statin use among patients with cirrhosis secondary to alcohol use disorder and chronic hepatitis B (HBV) and C viruses, a Taiwanese cohort study was performed. When it comes to the reduction in decompensation rates with statin use, this was statistically significant for cirrhosis secondary to HBV (adjusted hazard ratio (aHR): 0.39) and HCV (aHR: 0.51) infections but not for cirrhosis secondary to alcohol use disorder (aHR: 0.69, confidence interval (CI): 0.47 - 1.07). When it comes to the reduction in liver-related mortality rates with statin use, this was only statistically significant for cirrhosis secondary to chronic HBV infection but not for cirrhosis secondary to chronic HCV infection or alcohol use disorder. As for the risk reduction in HCC development with statin use, this was borderline significant for cirrhosis secondary to chronic HCV but not for cirrhosis secondary to chronic HBV infection or alcohol use disorder [67]. In addition to the above findings, the above-mentioned study demonstrated a dose-dependent reduction in the risk of decompensation, mortality, and development of HCC with statin use.
Retrospective and prospective studies evaluating the impact of statins on CLD progression
Effects of statins on PHTN
Hepatic venous pressure gradient (HVPG) measurement is the best available method to evaluate the presence and severity of PHTN [68]. Human research has shed light on the reduction of hepatic resistance occurring with the use of simvastatin in cirrhotic patients via the enhancement of NO production and hepatosplenic shunting.
1) Evidence from a cohort study
In one study performed on 30 cirrhotic patients, the use of 40 mg oral simvastatin resulted in acute hemodynamic effects in the liver, with a decrease in hepatic vascular resistance (-14%) and an increase in hepatic blood flow (+20%), only around 30 min after the time of administration. This reduction in hepatic vascular resistance was not associated with a decrease in portal pressure, however, due to concomitant increase in hepatic blood flow from an increase in NO products only in the hepatic vein. As a result, simvastatin was not shown to have a direct therapeutic potential in reducing cirrhosis decompensation in this study [69].
2) Evidence from randomized clinical trials
In the setting of the above evidence showing an increase in NO products with the use of statins and in the setting of evidence showing that supplementing NO enhances the effect of beta blockers on PHTN, further research was done to evaluate whether adding statins to beta blockers can enhance the latter’s effect on portal pressure. Current data suggest that beta blockers achieve their HVPG-lowering effects via a decrease in portal inflow, while statins attain theirs via a decrease in hepatic resistance.
A multicenter randomized control trial (RCT) was performed on 59 Child-Pugh A and B cirrhotic patients with severe PHTN from three Spanish hospitals. The effect of continuous oral administration of escalating doses of simvastatin (20 mg once daily for the first 14 days followed by 40 mg daily for the remaining 14 days) versus placebo on HVPG was studied over a period of 1 month. Compared to placebo, simvastatin use for a month led to the amelioration of hepatic perfusion and function as indicated by enhanced indocyanine clearance, without affecting systemic hemodynamics or increasing adverse event rates. In the experimental group, there was a moderate reduction (8.3%) in the HVPG, and around 30% of patients reached the treatment target of a 20% reduction from baseline or normalization of HVPG. Interestingly, this reduction in HVPG was observed whether patients were on beta-blockers (11%) or not (5.9%), and the degree of reduction was higher in patients concomitantly taking simvastatin and beta-blockers [70].
Results of these studies were supported by another proof-of-concept study that compared the effects of a 1-month use of atorvastatin (daily oral dose of 20 mg)-propranolol combination therapy with that of propranolol monotherapy on HVPG reduction. Although results revealed a greater degree of reduction in HVPG among patients in the atorvastatin-propranolol combination arm compared to that seen in the propranolol monotherapy one, there was no major difference between the two arms in rates of variceal bleeds, hepatic encephalopathy, endoscopic variceal ligation, SBP, need for paracentesis, and death. As a result, these results suggest but do not confirm a therapeutic potential of the combination therapy in cirrhotic patients [71].
In order to assess whether prolonging simvastatin treatment to more than a 1-month period has equivalent or superior effects on PHTN to that administered over a period of 1-month, further studies were designed and performed with longer follow-up time. A blinded RCT was performed on 24 Child-Pugh A and B cirrhotic patients, 67% of which were on non-selective beta-blockers for medium or large esophageal varices and 30% of which have had a history of previous variceal bleeding at time of study initiation. The effect of continuous oral administration of 40 mg simvastatin versus placebo on hepatic portal pressure was studied over a period of 3 months. A significant decrease in HVPG (-15%) was noted in the experimental group versus no change in the placebo group. Multivariate regression analysis revealed that this reduction in HVPG in the experimental group was more pronounced in patients with medium and large esophageal varices and those with a history of variceal bleeding than in those without these PHTN complications. Interestingly, however, bivariate analysis failed to show an association between the severity of PHTN and the hemodynamic response to simvastatin. This discrepancy was attributed to the limitations related to the small sample size. As such, while these results suggest that simvastatin can lower portal pressure with its hemodynamic effect being more pronounced in patients with severe PHTN, further studies with larger sample size are needed to confirm the results. Of note, compared to placebo, simvastatin was associated with some improvement in liver function in the absence of any significant increase in adverse events, which further encourages evaluation of simvastatin in future studies [72].
In order to check whether this reduction in HVPG would reflect in a reduction in rebleeding rates in cirrhotic patients with esophageal varices, a recent multicenter double blinded RCT was performed. This RCT included 158 cirrhotic patients with a history of variceal bleed. Its aim was to compare cirrhosis progression, rebleeding, and mortality rates after the addition of 40 mg simvastatin to the standard variceal bleed therapy (beta-blockers and esophageal band ligation) over a follow-up period of 24 months. Results showed no difference between rebleeding rates among the experimental and control groups (P = 0.58). Similarly, there was no difference when it came to cirrhosis complications, including the development of ascites, SBP, HRS, or portal vein thrombosis among both groups [73]. Despite the lack of reduction in variceal rebleeding and cirrhosis complication rates, simvastatin was shown to increase survival rates when added to the standard variceal bleed therapy in Child-Pugh A and B but not C patients. Although the adverse event rate in the simvastatin arm was not superior to that in the placebo arm, the fact that two patients with severe cirrhosis who belonged to the experimental group developed rhabdomyolysis raises safety concerns about the use of statins in advanced cirrhosis. The results of this trial are not generalizable to all patients with cirrhosis who recover from a variceal bleed because those with creatinine > 2 mg/dL, advanced liver dysfunction with a Child-Pugh score above 13, and complete portal vein thrombosis were excluded. In addition, the assessment of the study hypothesis might have been underpowered due to the fact that 11 patients were excluded from the analysis. Moreover, the lack of data on the potential impact of alcohol abstinence on the study outcomes is another limitation.
In the setting of conflicting results and questionable safety concerns of statins in advanced cirrhosis, stronger and more reliable data from further prospective trials are needed. In an ongoing phase III prospective multicenter double blind RCT, called the SACRED trial (NCT 03654053), patients with compensated cirrhosis and PHTN from 11 VA Medical Centers are stratified based upon the concomitant use of nonselective beta-blockers and then randomized to 40 mg simvastatin versus placebo over a follow-up period of 24 months. Participants will be observed for the development of hepatic decompensation (variceal hemorrhage, ascites, encephalopathy), HCC, liver-related death, death from any cause, and/or complications of statin therapy.
Use of statins in NAFLD and NASH
At present, NAFLD constitutes the most common liver disease [74]. In patients with visceral obesity, insulin resistance, and metabolic syndrome, there is a high propensity for NAFLD to progress to NASH and then to cirrhosis [75]. Since there are no labeled therapeutic agents that have been approved for patients with NAFLD/NASH, and since animal NASH models have reflected improvements in liver histology following exposure to statins, several studies have evaluated the use of statins in NAFLD/NASH patients.
1) Evidence from cohort studies
In one European study performed on 1,201 patients with suspected NASH, liver biopsies performed on the 107 patients who were receiving at least 6 months of statins (mainly moderate-intensity simvastatin) were found to have significantly lower rates of steatosis (odds ratio (OR): 0.48, 95% CI: 0.26 - 0.94), NASH (OR: 0.62, 95% CI: 0.40 - 0.97), and advanced fibrosis (fibrosis stage F2 - F4; OR: 0.59, 95% CI: 0.34 - 0.98) compared to those performed on the remaining patients not on statins [76].
In another prospective study, 20 patients with steatohepatitis received rosuvastatin over a 12-months period, and liver biopsies were performed at the end of the study to assess the degree of liver fibrosis. Interestingly, liver biopsies from 19 out of the 20 patients revealed regression of the steatohepatitis present at baseline. Since this study had no control group and since the degree of hepatic fibrosis was neither graded at baseline nor at the end of the study, the interpretation of these promising results remains difficult [77].
As such, low-quality evidence has thus far supported the idea that statin treatment in NAFLD/NASH patients tend to improve liver fibrosis, impede disease progression, and improve cardiovascular morbidity and mortality rates. Nevertheless, conclusions cannot be drawn before these promising results are confirmed by careful prospective RCTs evaluating baseline versus on-trial statin use as a possible confounder in NAFLD/NASH patients.
2) Evidence from randomized clinical trials
Several RCTs evaluating the therapeutic potential of statins in NAFLD/NASH patients have been designed since this population is at high risk of cardiovascular events and using statins that might offer substantial hepatic and cardiovascular-related adverse event reduction could be of therapeutic potential. Although both atorvastatin and simvastatin have been studies, promising evidence exists supporting the use of atorvastatin in not only preventing cardiovascular events but also in slowing liver disease progression.
When it comes to the use of atorvastatin in NAFLD/NASH patients, a post hoc analysis of three large RCTs including more than 11,000 NAFLD/NASH patients revealed a reduction in liver enzymes and an improvement in liver fat infiltration with atorvastatin use. Compared to NAFLD/NASH patients not receiving statin, and to statin-treated patients with normal livers, NAFLD/NASH patients who were treated with statins had a 50% reduction in cardiovascular disease morbidity and mortality [78].
Supporting evidence regarding the use of atorvastatin in NAFLD/NASH comes from a prospective open-label randomized study performed on 186 non-diabetic patients with both biochemical and ultrasonographic evidence of NAFLD at baseline. Patients were randomly allocated to receive atorvastatin 20 mg per day, fenofibrate 200 mg per day, or the combination of both drugs over a 54-week period. At the end of the follow-up period, a significantly higher percentage of patients receiving atorvastatin or combination therapy no longer had evidence of NAFLD after 54 weeks of treatment compared to the fenofibrate group with a P value of < 0.009. Sixty-seven percent of the patients receiving atorvastatin, 42% of those receiving fenofibrate, and 70% of those receiving the combination therapy no longer had biochemical and ultrasonographic evidence of NAFLD with a significant P value of < 0.05 compared to baseline. Interestingly, however, this statistical significance of the superiority of atorvastatin over fenofibrate did not exist before combining the two outcomes (biochemical versus ultrasonographic evidence of NAFLD) into one [79]. A limitation of this study was that it did not address the primary outcomes of all-cause mortality and liver-related morbidity.
When it comes to the use of simvastatin in NAFLD/NASH, a double-blinded randomized placebo-controlled trial was designed to assess the effects of simvastatin versus placebo in 16 participants with biopsy-proven NASH over a 12-month period. Serum aminotransferases and repeated liver biopsies were used to assess improvement. Although there was a 26% reduction in low-density lipoprotein within the simvastatin group compared with placebo, no statistically significant improvement in serum aminotransferases, hepatic steatosis, or stage of fibrosis existed between both groups [80]. A limitation of this study was that it did not address the primary outcomes of all-cause mortality and liver-related morbidity. It also had a modest statistical power due to being only a pilot trial.
Use of statins and the risk of development of HCC in CLD and cirrhosis
Several studies have shed light on the idea that the use of statins in patients with CLD could be protective against the development of HCC [81]. Many hypotheses have been developed regarding the mechanisms, through which statins convey their protective effects against HCC development in CLD. Suggested mechanisms include chemoprevention, inhibition of CLD progression to cirrhosis, and impedance of cirrhosis evolution and decompensation [82].
1) Evidence from cohort studies
In an early retrospective study, patients on statins were shown to have a dose-related reduction in the rates of HCC as compared to a matched control from the US population [83]. In a subsequent cohort study that included 27,883 HCV patients with HCC, this dose-related reduction in the rate of HCC development was also confirmed. Cumulative daily statin doses of 28 - 89mg, 90 - 180mg, and 180 mg were shown to result in adjusted HRs of 0.66 (95% CI: 0.59 - 0.74), 0.47 (95% CI: 0.40 - 0.56), and 0.33 (95% CI: 0.25 - 0.42) to develop HCC [84]. Based on data from a recent meta-analysis of 10 studies that included a total of 1.4 million patients, among which 4,298 cases had HCC, the number of East Asian patients needed to be treated with statin to prevent one case of HCC per year was 5,209. In addition, the number of patients with HBV-related cirrhosis needed to be treated with statin to prevent one case of HCC per year was 57 [85].
A prospective propensity score-matched cohort study included 16,668 nationwide Swedish adults with viral hepatitis over the period extending from 2005 to 2013. Out of the 16,668 patients, 6,554, 1,780, and 8,334 patients were on lipophilic, hydrophilic, and no statins, respectively. Lipophilic statin users were found to have a significantly lower 10-year HCC risk compared to hydrophilic statin users. This risk reduction with the use of lipophilic statins was shown to be dose dependent. The study, however, was limited by the lack of lipid, fibrosis, or HCC surveillance data. As a result, further research is needed to evaluate whether the use of lipophilic statins can have a role in HCC prevention [86].
2) Evidence from randomized clinical trials
Although cohort studies have commented on the chemopreventive effects of lipophilic versus hydrophilic statins against HCC development, the designed RCTs to date have evaluated the use of statins in patients with an established diagnosis of HCC. As such, there are not RCTs to comment on the efficacy of statin in reducing HCC development.
When it comes to the use of lipophilic statins, a randomized placebo-controlled study (NCT03275376) was designed to compare the use of atorvastatin 10 mg versus placebo in terms of overall survival, tumor response, and progression-free survival in patients receiving sorafenib therapy for advanced HCC in the Taichung Veterans General Hospital. This trial was terminated due to slow patient enrollment.
When it comes to the use of hydrophilic statins, such as pravastatin, in HCC subjects, there was a recent non-blind RCT (PRODIGE-11/FFCD 0803) that was designed to compare the 40 mg sorafenib monotherapy with pravastatin-sorafenib combination therapy in 323 patients with Child-Pugh A cirrhosis and advanced HCC. This trial, too, had to be terminated at interim analysis for proof of lack of efficacy of the latter arm. Although pravastatin addition was safe, no differences in terms of survival (HR = 1.00) or progression-free survival (HR = 1.00) existed between both groups. Data from an a multicentric prospective non-comparative RCT (PRODIGE-21) performed on patients with Child-Pugh B and advanced HCC revealed a shorter median survival with pravastatin 40 mg daily alone (3.1 months), as compared with either sorafenib monotherapy (3.8 months), sorafenib and pravastatin combination (4 months) and standard of care (3.5 months) [87].
| Conclusions and Future Directions | ▴Top |
In summary, several lines of evidence have led to a change in the pattern of statin use in patients with CLD or cirrhosis. While early data depicted statins as harmful agents for the liver, current evidence from preclinical and clinical studies suggests that they might have positive impact on CLD or cirrhosis. Low-quality evidence supports the fact that statins reduce mortality in CLD. Moderate-quality evidence suggests that statins reduce the risk of hepatic decompensation, variceal bleeding, and mortality, especially among patients with compensated cirrhosis. Combining this data with the long track-record of safety and tolerability of statins and their potential benefits in HCC risk reduction, hepatologists might soon rely on statins to achieve better outcomes in their CLD or cirrhosis patients without significant additional costs.
Although most of the above-mentioned studies have been through adjustments for potential confounders, it is likely that their results might have overestimated the potential benefits of statins due to possible residual confounding in statin users. The potential biases in observational studies have been widely recognized and likely explain the low rate of transportability of results from observational studies to RCTs [88]. As such, additional large prospective interventional RCTs are needed to be designed and carried out to better evaluate the association between statin exposure and the risk of fibrosis progression and development of cirrhosis in patients with non-cirrhotic CLDs, the risk of progression of PHTN in patients with cirrhosis, and the mortality rates in patients with cirrhotic or non-cirrhotic CLDs.
Acknowledgments
The completion of this research paper could not have been possible without the participation and assistance of so many people, and their contributions are sincerely appreciated and gratefully acknowledged. I would like to express my deep appreciation to all the team members that worked hard on literature review and writing the manuscript, with special thanks to Dr. Liliane Deeb for her supervision and guidance.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Author Contributions
Literature review: Malek Kreidieh and Rachelle Hamadi. Manuscript writing: Malek Kreidieh and Rachelle Hamadi. Manuscript revision and work supervision and guidance: Liliane Deeb, Mira Alsheikh, and Hassan Al Moussawi. All authors approved the final version of the manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
ALT: alanine transaminase; ApoE: apolipoprotein E; CLD: chronic liver disease; CCL2: dichloromethylene; CCR2: C-C motif chemokine receptor 2; eNOS: endothelial nitric oxide synthase; HBC: hepatitis B virus; HCC: hepatocellular carcinoma; HCV: hepatitis C virus; Hh: Hedgehog; HMG-CoA: 3-hydroxy-3-methylglutaryl coenzyme A; HPS: hepatopulmonary syndrome; HRS: hepatorenal syndrome; HSC: hepatic stellate cell; HVPG: hepatic venous pressure gradient; IL: interleukin; IFN: interferon; KLF2: Kruppel-like factor 2; LSEC: liver sinusoidal endothelial cell; MCDD: methionine-choline-deficient diet; MELD: model for end-stage liver disease; NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; NO: nitric oxide; PHTN: portal hypertension; PPARα: peroxisomal proliferator-activated receptor α; RCT: randomized control trial; RhoA: Ras homolog gene family member A; SBP: spontaneous bacterial peritonitis; TNF-α: tumor necrosis factor-α
| References | ▴Top |
- GBD 2015 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1603-1658.
doi - G. B. D. Cirrhosis Collaborators. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5(3):245-266.
doi - de Franchis R, Primignani M. Natural history of portal hypertension in patients with cirrhosis. Clin Liver Dis. 2001;5(3):645-663.
doi - Bosch J, Garcia-Pagan JC. Complications of cirrhosis. I. Portal hypertension. J Hepatol. 2000;32(1 Suppl):141-156.
doi - Tandon P, Garcia-Tsao G. Portal hypertension and hepatocellular carcinoma: prognosis and beyond. Clin Gastroenterol Hepatol. 2006;4(11):1318-1319.
doi pubmed - Ripoll C, Groszmann R, Garcia-Tsao G, Grace N, Burroughs A, Planas R, Escorsell A, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology. 2007;133(2):481-488.
doi pubmed - Grace ND, Groszmann RJ, Garcia-Tsao G, Burroughs AK, Pagliaro L, Makuch RW, Bosch J, et al. Portal hypertension and variceal bleeding: an AASLD single topic symposium. Hepatology. 1998;28(3):868-880.
doi pubmed - Garcia-Tsao G, Bosch J, Groszmann RJ. Portal hypertension and variceal bleeding—unresolved issues. Summary of an American Association for the study of liver diseases and European Association for the study of the liver single-topic conference. Hepatology. 2008;47(5):1764-1772.
doi pubmed - Benoit JN, Womack WA, Hernandez L, Granger DN. "Forward" and "backward" flow mechanisms of portal hypertension. Relative contributions in the rat model of portal vein stenosis. Gastroenterology. 1985;89(5):1092-1096.
doi - Whipple AO. The problem of portal hypertension in relation to the hepatosplenopathies. Ann Surg. 1945;122(4):449-475.
doi pubmed - Atkinson M, Losowsky MS. The mechanism of ascites formation in chronic liver disease. Q J Med. 1961;30:153-166.
- Sherlock S, Shaldon S. The aetiology and management of ascites in patients with hepatic cirrhosis: a review. Gut. 1963;4:95-105.
doi pubmed - Chou R, Dana T, Blazina I, Daeges M, Jeanne TL. Statins for prevention of cardiovascular disease in adults: evidence report and systematic review for the US preventive services task force. JAMA. 2016;316(19):2008-2024.
doi pubmed - Shalev V, Chodick G, Silber H, Kokia E, Jan J, Heymann AD. Continuation of statin treatment and all-cause mortality: a population-based cohort study. Arch Intern Med. 2009;169(3):260-268.
doi pubmed - Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, Blumenthal R, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet. 2016;388(10059):2532-2561.
doi - Blaha MJ, Martin SS. How do statins work?: changing paradigms with implications for statin allocation. J Am Coll Cardiol. 2013;62(25):2392-2394.
doi pubmed - Vargas JI, Arrese M, Shah VH, Arab JP. Use of statins in patients with chronic liver disease and cirrhosis: current views and prospects. Curr Gastroenterol Rep. 2017;19(9):43.
doi pubmed - Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. 2017;120(1):229-243.
doi pubmed - Salerno F, Gerbes A, Gines P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56(9):1310-1318.
- Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rodes J. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8(5):1151-1157.
doi pubmed - Sanyal AJ, Bosch J, Blei A, Arroyo V. Portal hypertension and its complications. Gastroenterology. 2008;134(6):1715-1728.
doi pubmed - Izadpanah R, Schachtele DJ, Pfnur AB, Lin D, Slakey DP, Kadowitz PJ, Alt EU. The impact of statins on biological characteristics of stem cells provides a novel explanation for their pleiotropic beneficial and adverse clinical effects. Am J Physiol Cell Physiol. 2015;309(8):C522-531.
doi pubmed - Musial J, Undas A, Gajewski P, Jankowski M, Sydor W, Szczeklik A. Anti-inflammatory effects of simvastatin in subjects with hypercholesterolemia. Int J Cardiol. 2001;77(2-3):247-253.
doi - Ridker PM, Rifai N, Lowenthal SP. Rapid reduction in C-reactive protein with cerivastatin among 785 patients with primary hypercholesterolemia. Circulation. 2001;103(9):1191-1193.
doi pubmed - Albert MA, Staggers J, Chew P, Ridker PM, Investigators P. The pravastatin inflammation CRP evaluation (PRINCE): rationale and design. Am Heart J. 2001;141(6):893-898.
doi pubmed - Lefer DJ. Statins as potent antiinflammatory drugs. Circulation. 2002;106(16):2041-2042.
doi pubmed - McGirt MJ, Lynch JR, Parra A, Sheng H, Pearlstein RD, Laskowitz DT, Pelligrino DA, et al. Simvastatin increases endothelial nitric oxide synthase and ameliorates cerebral vasospasm resulting from subarachnoid hemorrhage. Stroke. 2002;33(12):2950-2956.
doi pubmed - Kalinowski L, Dobrucki LW, Brovkovych V, Malinski T. Increased nitric oxide bioavailability in endothelial cells contributes to the pleiotropic effect of cerivastatin. Circulation. 2002;105(8):933-938.
doi pubmed - Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97(12):1129-1135.
doi pubmed - Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem. 1997;272(50):31725-31729.
doi pubmed - Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, Sanchez-Pascuala R, Hernandez G, Diaz C, Lamas S. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest. 1998;101(12):2711-2719.
doi pubmed - O'Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation. 1997;95(5):1126-1131.
doi pubmed - Zaccherini G, Tufoni M, Bernardi M, Caraceni P. Prevention of cirrhosis complications: looking for potential disease modifying agents. J Clin Med. 2021;10(19):4590.
doi pubmed - Bays H, Cohen DE, Chalasani N, Harrison SA, The National Lipid Association's Statin Safety Task F. An assessment by the Statin Liver Safety Task Force: 2014 update. J Clin Lipidol. 2014;8(3 Suppl):S47-57.
doi pubmed - Schierwagen R, Uschner FE, Magdaleno F, Klein S, Trebicka J. Rationale for the use of statins in liver disease. Am J Physiol Gastrointest Liver Physiol. 2017;312(5):G407-G412.
doi pubmed - Gracia-Sancho J, Russo L, Garcia-Caldero H, Garcia-Pagan JC, Garcia-Cardena G, Bosch J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut. 2011;60(4):517-524.
doi pubmed - Trebicka J, Schierwagen R. Statins, Rho GTPases and KLF2: new mechanistic insight into liver fibrosis and portal hypertension. Gut. 2015;64(9):1349-1350.
doi pubmed - Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA, Jr., Garcia-Cardena G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280(29):26714-26719.
doi pubmed - Marrone G, Russo L, Rosado E, Hide D, Garcia-Cardena G, Garcia-Pagan JC, Bosch J, et al. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. J Hepatol. 2013;58(1):98-103.
doi pubmed - Marrone G, Maeso-Diaz R, Garcia-Cardena G, Abraldes JG, Garcia-Pagan JC, Bosch J, Gracia-Sancho J. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: behind the molecular mechanisms of statins. Gut. 2015;64(9):1434-1443.
doi pubmed - Trebicka J, Hennenberg M, Laleman W, Shelest N, Biecker E, Schepke M, Nevens F, et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology. 2007;46(1):242-253.
doi pubmed - Chong LW, Hsu YC, Lee TF, Lin Y, Chiu YT, Yang KC, Wu JC, et al. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. 2015;15:22.
doi pubmed - Russo L, Gracia-Sancho J, Garcia-Caldero H, Marrone G, Garcia-Pagan JC, Garcia-Cardena G, Bosch J. Addition of simvastatin to cold storage solution prevents endothelial dysfunction in explanted rat livers. Hepatology. 2012;55(3):921-930.
doi pubmed - Klein S, Klosel J, Schierwagen R, Korner C, Granzow M, Huss S, Mazar IG, et al. Atorvastatin inhibits proliferation and apoptosis, but induces senescence in hepatic myofibroblasts and thereby attenuates hepatic fibrosis in rats. Lab Invest. 2012;92(10):1440-1450.
doi pubmed - Trebicka J, Hennenberg M, Odenthal M, Shir K, Klein S, Granzow M, Vogt A, et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J Hepatol. 2010;53(4):702-712.
doi pubmed - Nie L, Imamura M, Itoh H, Ueno H. Pitavastatin enhances the anti-fibrogenesis effects of candesartan, an angiotensin II receptor blocker, on CCl4-induced liver fibrosis in rats. J UOEH. 2004;26(2):165-177.
doi pubmed - El-Ashmawy NE, El-Bahrawy HA, Shamloula MM, Ibrahim AO. Antifibrotic effect of AT-1 blocker and statin in rats with hepatic fibrosis. Clin Exp Pharmacol Physiol. 2015;42(9):979-987.
doi pubmed - Shirin H, Sharvit E, Aeed H, Gavish D, Bruck R. Atorvastatin and rosuvastatin do not prevent thioacetamide induced liver cirrhosis in rats. World J Gastroenterol. 2013;19(2):241-248.
doi pubmed - Rodriguez S, Raurell I, Torres-Arauz M, Garcia-Lezana T, Genesca J, Martell M. A nitric oxide-donating statin decreases portal pressure with a better toxicity profile than conventional statins in cirrhotic rats. Sci Rep. 2017;7:40461.
doi pubmed - Abraldes JG, Rodriguez-Vilarrupla A, Graupera M, Zafra C, Garcia-Caldero H, Garcia-Pagan JC, Bosch J. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J Hepatol. 2007;46(6):1040-1046.
doi pubmed - Hsu SJ, Wang SS, Hsin IF, Huang HC, Lee FY, Lee JY, Lin HC, et al. Effects of simvastatin on the portal-systemic collateral vascular response to endothelin-1 and shunting degree in portal hypertensive rats. Scand J Gastroenterol. 2013;48(7):831-838.
doi pubmed - Huang HC, Wang SS, Lee JY, Chen YC, Lee FY, Lin HC, Chang CC, et al. Simvastatin effects on portal-systemic collaterals of portal hypertensive rats. J Gastroenterol Hepatol. 2010;25(8):1401-1409.
doi pubmed - Uschner FE, Ranabhat G, Choi SS, Granzow M, Klein S, Schierwagen R, Raskopf E, et al. Statins activate the canonical hedgehog-signaling and aggravate non-cirrhotic portal hypertension, but inhibit the non-canonical hedgehog signaling and cirrhotic portal hypertension. Sci Rep. 2015;5:14573.
doi pubmed - Arab JP, Shah VH. Statins and portal hypertension: A tale of two models. Hepatology. 2016;63(6):2044-2047.
doi pubmed - Chang CC, Wang SS, Huang HC, Lee JY, Lee FY, Lin HC, Lee SD. Pravastatin administration does not induce detrimental effects on hemodynamics and collaterals of portal hypertensive rats. J Gastroenterol Hepatol. 2010;25(8):1394-1400.
doi pubmed - Nseir W, Mahamid M. Statins in nonalcoholic fatty liver disease and steatohepatitis: updated review. Curr Atheroscler Rep. 2013;15(3):305.
doi pubmed - Park HS, Jang JE, Ko MS, Woo SH, Kim BJ, Kim HS, Park HS, et al. Statins increase mitochondrial and peroxisomal fatty acid oxidation in the liver and prevent non-alcoholic steatohepatitis in mice. Diabetes Metab J. 2016;40(5):376-385.
doi pubmed - Schierwagen R, Maybuchen L, Hittatiya K, Klein S, Uschner FE, Braga TT, Franklin BS, et al. Statins improve NASH via inhibition of RhoA and Ras. Am J Physiol Gastrointest Liver Physiol. 2016;311(4):G724-G733.
doi pubmed - Wu W, Zhao L, Yang P, Zhou W, Li B, Moorhead JF, Varghese Z, et al. Inflammatory Stress Sensitizes the Liver to Atorvastatin-Induced Injury in ApoE-/- Mice. PLoS One. 2016;11(7):e0159512.
doi pubmed - Okada Y, Yamaguchi K, Nakajima T, Nishikawa T, Jo M, Mitsumoto Y, Kimura H, et al. Rosuvastatin ameliorates high-fat and high-cholesterol diet-induced nonalcoholic steatohepatitis in rats. Liver Int. 2013;33(2):301-311.
doi pubmed - Kamada Y, Kiso S, Yoshida Y, Chatani N, Kizu T, Hamano M, Egawa M, et al. Pitavastatin ameliorated the progression of steatohepatitis in ovariectomized mice fed a high fat and high cholesterol diet. Hepatol Res. 2013;43(4):401-412.
doi pubmed - Yang YH, Chen WC, Tsan YT, Chen MJ, Shih WT, Tsai YH, Chen PC. Statin use and the risk of cirrhosis development in patients with hepatitis C virus infection. J Hepatol. 2015;63(5):1111-1117.
doi pubmed - Huang YW, Lee CL, Yang SS, Fu SC, Chen YY, Wang TC, Hu JT, et al. Statins reduce the risk of cirrhosis and its decompensation in chronic hepatitis B patients: a nationwide cohort study. Am J Gastroenterol. 2016;111(7):976-985.
doi pubmed - Kumar S, Grace ND, Qamar AA. Statin use in patients with cirrhosis: a retrospective cohort study. Dig Dis Sci. 2014;59(8):1958-1965.
doi pubmed - Mohanty A, Tate JP, Garcia-Tsao G. Statins are associated with a decreased risk of decompensation and death in veterans with hepatitis C-related compensated cirrhosis. Gastroenterology. 2016;150(2):430-440.e431.
doi pubmed - Motzkus-Feagans C, Pakyz AL, Ratliff SM, Bajaj JS, Lapane KL. Statin use and infections in Veterans with cirrhosis. Aliment Pharmacol Ther. 2013;38(6):611-618.
doi pubmed - Chang FM, Wang YP, Lang HC, Tsai CF, Hou MC, Lee FY, Lu CL. Statins decrease the risk of decompensation in hepatitis B virus- and hepatitis C virus-related cirrhosis: A population-based study. Hepatology. 2017;66(3):896-907.
doi pubmed - Suk KT. Hepatic venous pressure gradient: clinical use in chronic liver disease. Clin Mol Hepatol. 2014;20(1):6-14.
doi pubmed - Zafra C, Abraldes JG, Turnes J, Berzigotti A, Fernandez M, Garca-Pagan JC, Rodes J, et al. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology. 2004;126(3):749-755.
doi pubmed - Abraldes JG, Albillos A, Banares R, Turnes J, Gonzalez R, Garcia-Pagan JC, Bosch J. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology. 2009;136(5):1651-1658.
doi pubmed - Bishnu S, Ahammed SM, Sarkar A, Hembram J, Chatterjee S, Das K, Dhali GK, et al. Effects of atorvastatin on portal hemodynamics and clinical outcomes in patients with cirrhosis with portal hypertension: a proof-of-concept study. Eur J Gastroenterol Hepatol. 2018;30(1):54-59.
doi pubmed - Pollo-Flores P, Soldan M, Santos UC, Kunz DG, Mattos DE, da Silva AC, Marchiori RC, et al. Three months of simvastatin therapy vs. placebo for severe portal hypertension in cirrhosis: A randomized controlled trial. Dig Liver Dis. 2015;47(11):957-963.
doi pubmed - Abraldes JG, Villanueva C, Aracil C, Turnes J, Hernandez-Guerra M, Genesca J, Rodriguez M, et al. Addition of simvastatin to standard therapy for the prevention of variceal rebleeding does not reduce rebleeding but increases survival in patients with cirrhosis. Gastroenterology. 2016;150(5):1160-1170.e1163.
- Cotter TG, Rinella M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology. 2020;158(7):1851-1864.
doi pubmed - Mirza MS. Obesity, Visceral Fat, and NAFLD: Querying the Role of Adipokines in the Progression of Nonalcoholic Fatty Liver Disease. ISRN Gastroenterol. 2011;2011:592404.
doi pubmed - Dongiovanni P, Petta S, Mannisto V, Mancina RM, Pipitone R, Karja V, Maggioni M, et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J Hepatol. 2015;63(3):705-712.
doi pubmed - Kargiotis K, Athyros VG, Giouleme O, Katsiki N, Katsiki E, Anagnostis P, Boutari C, et al. Resolution of non-alcoholic steatohepatitis by rosuvastatin monotherapy in patients with metabolic syndrome. World J Gastroenterol. 2015;21(25):7860-7868.
doi pubmed - Athyros VG, Boutari C, Stavropoulos K, Anagnostis P, Imprialos KP, Doumas M, Karagiannis A. Statins: an under-appreciated asset for the prevention and the treatment of NAFLD or NASH and the related cardiovascular risk. Curr Vasc Pharmacol. 2018;16(3):246-253.
doi pubmed - Athyros VG, Mikhailidis DP, Didangelos TP, Giouleme OI, Liberopoulos EN, Karagiannis A, Kakafika AI, et al. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: a randomised study. Curr Med Res Opin. 2006;22(5):873-883.
doi pubmed - Nelson A, Torres DM, Morgan AE, Fincke C, Harrison SA. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: A randomized placebo-controlled trial. J Clin Gastroenterol. 2009;43(10):990-994.
doi pubmed - Bosch J, Gracia-Sancho J, Abraldes JG. Cirrhosis as new indication for statins. Gut. 2020;69(5):953-962.
doi pubmed - Gunarathne LS, Rajapaksha H, Shackel N, Angus PW, Herath CB. Cirrhotic portal hypertension: from pathophysiology to novel therapeutics. World J Gastroenterol. 2020;26(40):6111-6140.
doi pubmed - Islam MM, Poly TN, Walther BA, Yang HC, Jack Li YC. Statin use and the risk of hepatocellular carcinoma: a meta-analysis of observational studies. Cancers (Basel). 2020;12(3):671.
doi pubmed - Tsan YT, Lee CH, Ho WC, Lin MH, Wang JD, Chen PC. Statins and the risk of hepatocellular carcinoma in patients with hepatitis C virus infection. J Clin Oncol. 2013;31(12):1514-1521.
doi pubmed - Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144(2):323-332.
doi pubmed - Simon TG, Duberg AS, Aleman S, Hagstrom H, Nguyen LH, Khalili H, Chung RT, et al. Lipophilic statins and risk for hepatocellular carcinoma and death in patients with chronic viral hepatitis: results from a nationwide Swedish population. Ann Intern Med. 2019;171(5):318-327.
doi pubmed - Blanc JF, Khemissa F, Bronowicki JP, Monterymard C, Perarnau JM, Bourgeois V, Obled S, et al. Phase 2 trial comparing sorafenib, pravastatin, their combination or supportive care in HCC with Child-Pugh B cirrhosis. Hepatol Int. 2021;15(1):93-104.
doi pubmed - Collins R, MacMahon S. Reliable assessment of the effects of treatment on mortality and major morbidity, I: clinical trials. Lancet. 2001;357(9253):373-380.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.