

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website https://www.gastrores.org |
Review
Volume 14, Number 2, April 2021, pages 49-65
Idiopathic Non-Cirrhotic Portal Hypertension and Porto-Sinusoidal Vascular Disease: Review of Current Data
Michel Kmeida, Xiuli Liub, Samuel Ballentinec, Hwajeong Leea, d ![]()
aDepartment of Pathology and Laboratory Medicine, Albany Medical Center, Albany, NY, USA
bDepartment of Pathology and Laboratory Medicine, University of Florida at Gainesville, FL, USA
cDepartment of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO, USA
dCorresponding Author: Hwajeong Lee, Department of Pathology and Laboratory Medicine, Albany Medical Center, 47 New Scotland Ave., MC81, Albany, NY 12208, USA
Manuscript submitted March 7, 2021, accepted March 30, 2021, published online April 21, 2021
Short title: INCPH and PSVD: Review of Current Data
doi: https://doi.org/10.14740/gr1376
- Abstract
- Introduction
- Background
- INCPH vs. PSVD
- Nomenclature
- Definitions
- Alternative Diagnoses Excluded From PSVD Spectrum Despite Overlapping Histomorphology
- Epidemiology
- Pathophysiology and Etiologic Associations
- Diagnosis
- Management
- Clinical Outcomes
- Summary
- References
| Abstract | ▴Top |
Idiopathic non-cirrhotic portal hypertension (INCPH) is a clinicopathologic disease entity characterized by the presence of clinical signs and symptoms of portal hypertension (PH) in the absence of liver cirrhosis or known risk factors accountable for PH. Multiple hematologic, immune-related, infectious, hereditary and metabolic risk factors have been associated with this disorder. Still, the exact etiopathogenesis is largely unknown. The recently proposed porto-sinusoidal vascular disease (PSVD) scheme broadens the spectrum of the disease by also including patients without clinical PH who are found to have similar histopathologic findings on core liver biopsies. Three histomorphologic lesions have been identified as specific for PSVD to include obliterative portal venopathy, nodular regenerative hyperplasia and incomplete septal cirrhosis/fibrosis. However, these findings are often subtle, under-recognized and subjective with low interobserver agreement among pathologists. Additionally, the natural history of the subclinical forms of the disease remains unexplored. The clinical course is more favorable compared to cirrhosis patients, especially in the absence of clinical PH or liver dysfunction. There are no universally accepted guidelines in regard to diagnosis and treatment of INCPH/PSVD. Hence, this review emphasizes the need to raise awareness of this entity by highlighting its complex pathophysiology and clinicopathologic associations. Lastly, formulation of standardized diagnostic criteria with clinical validation is necessary to avoid misclassifying vascular diseases of the liver and to develop and implement targeted therapeutic strategies.
Keywords: Liver; Cirrhosis; Nodular regenerative hyperplasia; Incomplete septal cirrhosis; Non-cirrhotic; Portal hypertension; Porto-sinusoidal; Vascular injury
| Introduction | ▴Top |
Idiopathic non-cirrhotic portal hypertension (INCPH) is a clinicopathologic disease entity wherein the patient presents with portal hypertension (PH) without cirrhosis or underlying liver disease (chronic viral hepatitis, fatty liver disease, autoimmune hepatitis, hereditary hemochromatosis, primary biliary cholangitis, congenital hepatic fibrosis, sarcoidosis, schistosomiasis, etc.) that can cause PH [1]. Over the past decade, there has been a growing recognition among gastroenterologists, hepatologists and pathologists that the histologic findings of INCPH are not entirely specific to those with PH, and similar changes can be found in patients without PH. Thus, the novel entity “porto-sinusoidal vascular disease (PSVD)” has been introduced to broaden the definitional spectrum of INCPH and capture pre-PH phase of INCPH as well as INCPH that is concurrent with other liver diseases [2]. Yet, understanding the concept and clinical significance of PSVD, especially without PH, remains elusive.
In this review paper, we aim to systematically review the currently available evidence and enhance our understanding of INCPH and PSVD. More specifically, we provide an overview of the definition, terminologies and epidemiology of INCPH/PSVD and review clinical, radiologic and histologic features of this rare entity, with emphasis on the evolving pathophysiologic understandings that are currently available. This review would lay out a solid foundation for future studies of the natural history of PSVD in patients with no PH at the time of diagnosis.
| Background | ▴Top |
PH is defined by the presence of an elevated hepatic venous pressure gradient (HVPG, the difference between free hepatic venous pressure and wedge hepatic venous pressure) exceeding 5 mm Hg. PH is often the result of increased resistance in the portal blood outflow tract [1]. Signs of PH include varices, porto-systemic collaterals, ascites and thrombocytopenia with splenomegaly (Table 1). Hepatic cirrhosis is the most common etiology of PH in the Western hemisphere while schistosomiasis is a frequent culprit in endemic areas to include southern and sub-Saharan Africa, South America, the Caribbean, parts of China and Southeast Asia [3-5]. The abnormal impedance to portal venous flow can occur at differing levels, such as the prehepatic (i.e., portal vein), intrahepatic (involvement of intraparenchymal sinusoids and venules) or post-hepatic (i.e., hepatic veins) level. When signs and symptoms of PH develop in the absence of liver cirrhosis, the condition is designated as non-cirrhotic portal hypertension (NCPH).
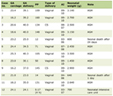 Click to view | Table 1. Symptoms and Signs of Portal Hypertension |
Interestingly, some chronic hepatopathies that ultimately culminate in the development of cirrhosis can manifest as PH even before liver cirrhosis develops and qualify for NCPH although admittingly, the terms NCPH and INCPH have been interchangeably used in the literature [5-7]. This includes alcoholic and nonalcoholic fatty liver disease (NAFLD), metabolic liver diseases and autoimmune diseases such as primary biliary cholangitis and primary sclerosing cholangitis. Exposure to medications and toxins, congenital hepatic fibrosis and infiltrative liver diseases (i.e., sarcoidosis) can also be involved in the pathogenesis of PH without cirrhosis [1, 3, 5].
Yet, some patients present with PH without identifiable cause including the entities mentioned above. Only after excluding cirrhosis by histologic examination, eliminating a mechanical (i.e., portal vein thrombosis, Budd-Chiari syndrome (BCS)) or physiologic (i.e., right-sided heart failure) blood flow obstruction in the portal and hepatic venous systems clinically and/or radiologically, and confirming the absence of chronic liver disease or known risk factors for PH, can a diagnosis of INCPH be rendered [1].
| INCPH vs. PSVD | ▴Top |
INCPH encompasses a heterogenous group of hepatic disorders for which the terminology was first introduced by Shouten et al in 2011 [1, 8]. Histologic features commonly seen in INCPH include obliterative portal venopathy (OPV), nodular regenerative hyperplasia and incomplete septal cirrhosis/fibrosis. Other findings include sinusoidal dilatation, mild perisinusoidal fibrosis, central vein abnormalities and variable portal vascular aberrancies (Table 2 [1, 2, 8-11]).
Click to view | Table 2. Histologic Features of Idiopathic Non-Cirrhotic Portal Hypertension/Porto-Sinusoidal Vascular Disease and Their Definitions [1, 2, 8-11] |
However, to date, this entity remains relatively poorly understood due to varying nomenclature and the lack of standardized diagnostic criteria. Furthermore, even though recent large-scale morphologic studies and collaborations on this topic [9, 12-14] enabled us to have a better understanding of the pathogenesis of this entity and recognize the wide histologic spectrum it displays, several shortcomings still exist.
Firstly, histologic features of INCPH are not entirely specific as they can be seen in liver specimens from patients without PH. Whether this finding represents a pre-PH phase of INCPH is currently unknown and this notion remains somewhat speculative [12]. For example, portal vascular aberrancies that have been described in INCPH were seen in “normal” aging livers [15] and liver samples from trauma patients [16]. Similarly, portal and lobular vascular changes that can be seen in INCPH were present in liver biopsies taken during gastric bypass surgery or cholecystectomy [13], in patients with hemochromatosis with or without history of phlebotomy [14] and in nine (69%) of 13 patients on dialysis [17], all in the absence of PH. Likewise, in a recent interobserver variability study, the control group patients without PH who showed some features of INCPH in their liver biopsies did not develop PH after a mean follow-up of 38 months [18]. In the absence of PH, these cases would not meet the diagnostic criteria for INCPH. Therefore, even if some of these indeed were to represent pre-PH phase of INCPH, we will not be able to diagnose these patients as having INCPH at the time of liver biopsy. Secondly, it has been recognized that certain liver conditions that must be excluded to establish a diagnosis of INCPH (i.e., fatty liver disease, viral and autoimmune hepatitis, among others) may in fact coexist with INCPH [2]. In such cases, the restrictive definition of INCPH would exclude these patients from this group.
These conceptual limitations have led the Vascular Liver Disease Interest Group (VALDIG) to propose a novel entity designed to broaden the whole aspects and spectrum of the disease in 2019. The term PSVD was introduced as the disease process primarily affects the hepatic sinusoids and portal venules [2].
However, this new classification is not without its own share of limitations. It might be too simplistic as it primarily relies on histomorphologic features for diagnosis which are known to be subtle and often under-recognized. Moreover, while this classification attempts to broaden its spectrum, it also excludes certain conditions that may well co-exist with PSVD from the definitional spectrum.
| Nomenclature | ▴Top |
Previously, different terminologies have been used for INCPH/PSVD. Idiopathic portal hypertension [19] and non-cirrhotic portal fibrosis [20] were commonly used in Eastern countries (Japan and India, respectively), while the terms “OPV” and “hepatoportal sclerosis” were endorsed in the Western literature [10]. Other terminologies that have been used for this entity include “benign intrahepatic portal hypertension”, “intrahepatic noncirrhotic portal hypertension”, “noncirrhotic portal hypertension”, “idiopathic noncirrhotic intrahepatic portal hypertension”, “partial nodular transformation”, “incomplete septal cirrhosis” and “nodular regenerative hyperplasia” [1-3, 6, 7, 10, 21]. Table 3 [3, 6, 12, 16, 19-68] summarizes the terminologies used for this entity.
Click to view | Table 3. Different Terminologies Used for Idiopathic Non-Cirrhotic Portal Hypertension/Porto-Sinusoidal Vascular Disease |
| Definitions | ▴Top |
In 2011, Shouten et al defined INCPH as a disease entity characterized by the presence of PH in the absence of hepatic fibrosis/cirrhosis or other factors that may be accountable for the development of PH [1, 8]. As a result, chronic liver diseases, portal, splenic and splanchnic venous thrombosis, and BCS must be first excluded to render the diagnosis [1, 8].
On the other hand, the diagnosis of PSVD requires a core liver biopsy of sufficient length (≥ 20 mm or with ≥ 10 portal tracts) with no evidence of cirrhosis and with either a specific sign of PH (varices, PH related bleeding, porto-systemic collaterals on imaging) or a specific histologic lesion (i.e., OPV, incomplete septal fibrosis or incomplete septal cirrhosis, nodular regenerative hyperplasia) for PSVD [2]. PSVD can also be diagnosed when not specific (of note, VALDIG used the particular term “not specific” in lieu of “nonspecific” in the manuscript) signs of PH (determined by VALDIG) and not specific histologic lesions of PSVD (determined by VALDIG) are present together, as long as a core ≥ 20 mm of non-cirrhotic liver is demonstrated [2]. As a result, in contrast to the INCPH scheme, neither the presence of a contributing factor for parenchymal liver disease (i.e., metabolic, viral or alcoholic liver disease) nor the absence of clinical PH excludes the diagnosis of PSVD if a specific histologic lesion is present in a non-cirrhotic liver. Additionally, portal vein thrombosis (particularly when it is not accompanied by cavernoma) does not eliminate the possibility of PSVD as it was found to be commonly encountered along the natural course of the disease [2].
Nevertheless, some entities are not to be included in the broad definitional spectrum of PSVD and, when present, should prompt an alternative diagnosis. Infiltrative liver diseases (i.e., sarcoidosis, malignancies), congenital hepatic fibrosis, schistosomiasis and heart failure must still be ruled out before diagnosing PSVD. Chronic cholestatic hepatopathies must also be excluded [2]. The following are conditions that closely resemble PSVD histologically and clinically, thus need to be excluded before rendering a PSVD diagnosis.
| Alternative Diagnoses Excluded From PSVD Spectrum Despite Overlapping Histomorphology | ▴Top |
BCS
BCS is characterized by the presence of hepatic venous outflow tract obstruction (HVOTO) in the absence of right-sided heart failure or constrictive pericarditis [69, 70]. The terms BCS and HVOTO can be used interchangeably. BCS can be primary when the obstruction in the hepatic venous tract is the result of a thrombus in the context of a hypercoagulable state or secondary when it is related to invasion or encasement by a malignant tumor or an abscess [69]. The obstruction can occur at any level between small intrahepatic veins and the right atrium including the suprahepatic inferior vena cava.
BCS is usually identified by noninvasive imaging modalities such as Doppler ultrasound, triphasic multidetector computed tomography (CT) and magnetic resonance imaging (MRI) [70]. In typical cases, a liver biopsy is not required as histologic findings fail to show independent prognostic value when adjusted for the model for end-stage liver disease (MELD) or Child-Pugh scores [71]. Liver biopsy may be performed when the obstruction is at the level of intrahepatic small venules thus is not detected on imaging, or when the patient presents with cirrhosis of unknown etiology. The biopsy shows features of venous outflow obstruction pattern injury, which is a known histologic mimic of PSVD [72, 73] (Fig. 1). Nevertheless, BCS/HVOTO is mainly a clinical/radiologic diagnosis and should not be considered a form of PSVD [2].
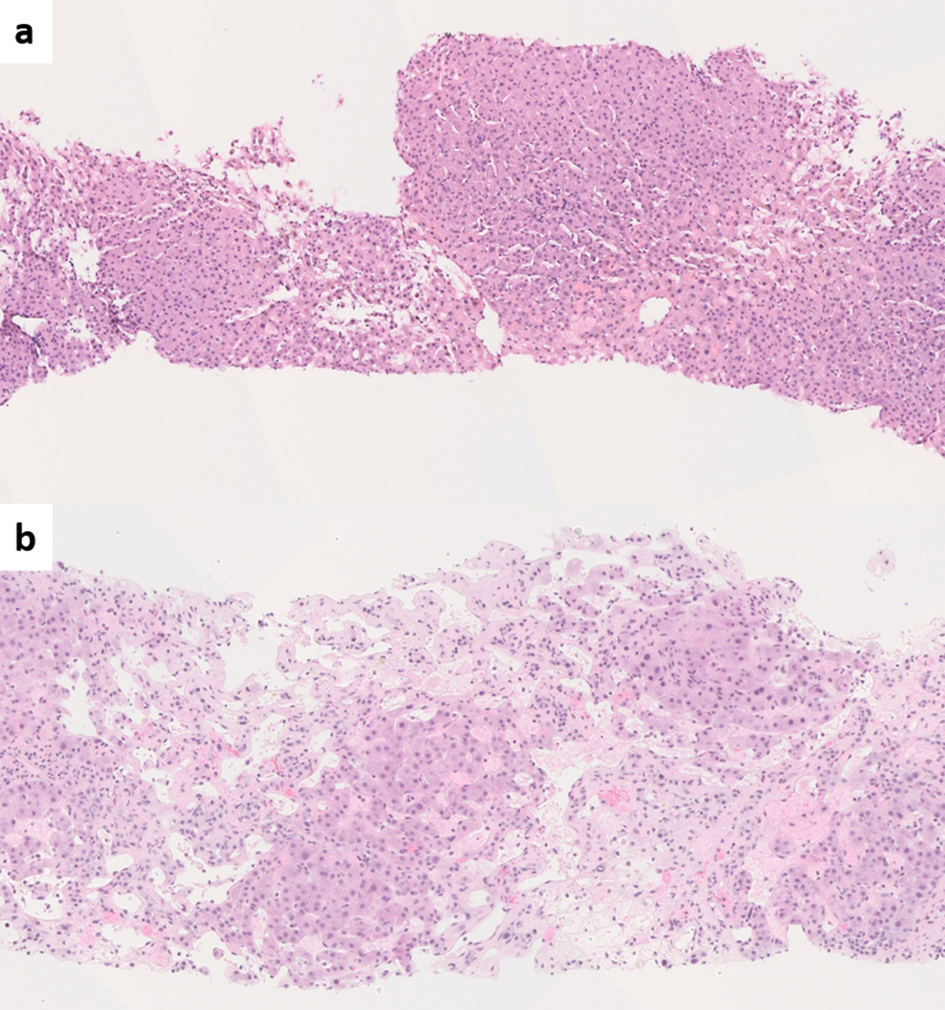 Click for large image | Figure 1. (a) Liver biopsy from Budd-Chiari syndrome (BCS) shows venous outflow obstruction pattern injury with mild centrizonal sinusoidal dilatation and congestion (hematoxylin and eosin (H&E), × 100). (b) Chronic BCS with extensive sinusoidal dilatation, centrizonal hepatocyte atrophy and dropout and fibrosis. The portal tracts are relatively spared (H&E, × 100). |
Sinusoidal obstruction syndrome (SOS)
SOS, previously known as a hepatic veno-occlusive disease (VOD), is regarded as a distinct entity that is not within the spectrum of PSVD. SOS is characterized by sinusoidal endothelial cell damage due to exposure to exogenous toxins that result in partial or complete occlusion of small intrahepatic veins [74]. SOS/VOD often occurs as a fatal complication following hematopoietic stem cell transplantation (HSCT), but it can also be seen after exposure to certain chemotherapeutic/immunomodulatory agents, in particular thiopurine derivatives [74]. The Seattle and Baltimore diagnostic clinical criteria have been extensively used in the setting of HSCT [75]. However, their applicability in non-HSCT patients is not well established, necessitating liver biopsy in some clinical settings [71, 73]. Common histologic findings include centrizonal congestion and fibrosis involving sinusoids and/or central veins [73] (Fig. 2). Again, these histologic features fall within the spectrum of venous outflow obstruction pattern injury. Hence, liver diseases in patients with known history of bone marrow transplantation are excluded from the definition of PSVD [2].
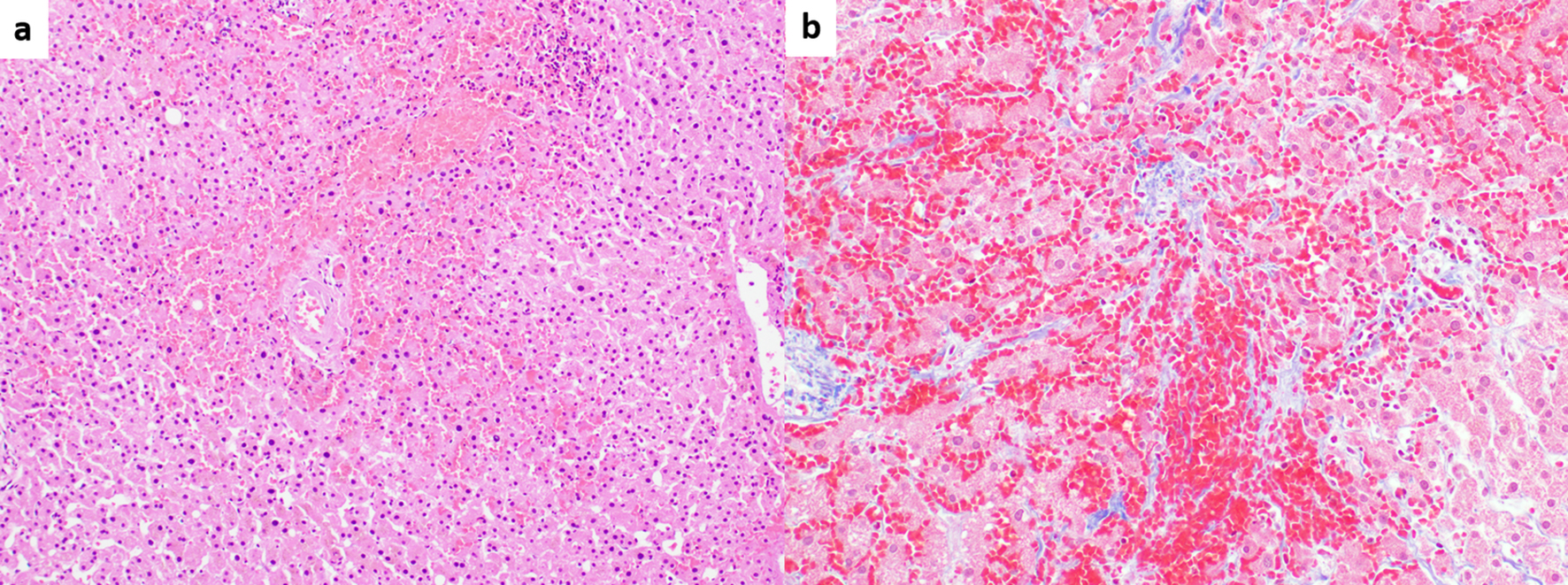 Click for large image | Figure 2. (a) Sinusoidal obstruction syndrome (SOS) associated with oxaliplatin chemotherapy. Marked centrizonal congestion and sinusoidal widening is noted (hematoxylin and eosin, × 100). (b) Trichrome stain shows perisinusoidal fibrosis (Masson-trichrome, × 200). |
Non-obstructive sinusoidal dilatation (NOSD)
NOSD has histomorphologic features that are overlapping with PSVD. NOSD is not universally defined; however, some authors define NOSD as a finding of sinusoidal lumina that are wider than one liver cell plate and present in multiple lobules with no artifactual tearing, in the absence of post-sinusoidal outflow block (PSOB) [76, 77]. Its clinical significance is unclear but the sinusoidal widening should not be explained by infiltration by sickle cells, hemophagocytic histiocytes, neoplasms, BCS, heart failure or small-for-size syndrome after liver transplantation, all of which feature PSOB. SOS/VOD should also be excluded even though NOSD can also be associated with drug exposures, notably oxaliplatin-based chemotherapy [77].
Peliosis
Peliosis is considered a severe form of sinusoidal dilatation with randomly distributed lobular cystic blood lakes [71]. Sinusoidal dilatation/peliosis are believed to be associated with an abnormal portal venous blood inflow or may represent a feature of severe systemic inflammatory reaction syndrome [77]. Peliosis, however, implicates a complete rupture of the reticulin framework which is not seen in NOSD [77]. PSVD may be part of the broader spectrum of sinusoidal dilatation/peliosis [73].
Congenital porto-systemic shunt (CPS)
CPS represents an abnormal intra- or extrahepatic communication between intestinal/splenic venous blood and systemic circulation bypassing liver parenchyma [78]. Extrahepatic CPS is referred to as Abernethy malformation and is excluded from the current definition of PSVD [2].
| Epidemiology | ▴Top |
It is difficult to estimate the incidence of INCPH/PSVD mainly due to varying/evolving nomenclature and geographic differences regarding the identification and classification of this disorder. Notably, PSVD encompasses a larger population compared to INCPH as it also includes patients without clinical PH. Moreover, PSVD was first introduced in 2019, thus the incidence of PSVD inclusive of those without PH is unknown.
INCPH has been widely recognized in Japan and the Indian subcontinent over the past few decades where it constitutes approximately 30-40% of PH cases [10, 22, 79]. However, due to increased awareness of the disease and changes in national health policy, the incidence of INCPH has dramatically decreased in Japan to a total number of 11 new cases per year [2]. The high incidence of INCPH in India appears to be related to less favorable socio-economic conditions [20, 80]. In North America and Europe, INCPH is considered a rare disease as it accounts for around 3-5% of PH cases [10]. However, its incidence is likely to be higher due to frequent under-recognition of this entity and the challenges inherent to the diagnosis and the interpretation of corresponding liver core biopsies. In fact, many INCPH patients might have been misdiagnosed as having cryptogenic or autoimmune cirrhosis especially in cases without previous liver biopsy [6].
Geographic demographic differences are also noted. Patients affected with INCPH skew significantly younger in India and Japan (age ranges of 30 - 49 years and 40 - 59 years, respectively) when compared to Western countries (age range 50 - 69 years) [2]. While the disease primarily affects women in Japan (although the gender gap is trending downward), it shows a male predilection in India, North America and Europe [2, 10]. INCPH has been rarely described in the pediatric population where it accounts for roughly 4.6% of all causes of PH [37]. In children, INCPH is more common in males and is more likely to be associated with an underlying malignancy or a genetic predisposition compared to adults [43, 45].
| Pathophysiology and Etiologic Associations | ▴Top |
The pathogenesis of INCPH/PSVD remains largely unknown but it is believed to be related to the injury to and occlusion of the intrahepatic portal microvasculature leading to increased resistance to portal blood flow at the presinusoidal level [7, 79, 81]. In cirrhosis-related PH, the portal venous bed decreases but the hepatic arterial bed increases leading to various intrahepatic shunts [82, 83]. In INCPH/PSVD, on the other hand, both venous and arterial beds diminish in size and intrahepatic shunts seldom occur [25, 27]. The attenuated portal venous system along with subsequent phlebosclerosis results in increased blood pressure in non-stenotic venous branches [7]. The ensuing abnormal/aberrant intrahepatic portal vasculature in INCPH has been extensively studied and classified by Ohbu et al [52]. In this study, a combination of phlebosclerosis of portal venous branches and dilated periportal sinusoids that are not communicating with the portal venous branches was commonly observed in INCPH and nodular regenerative hyperplasia. Phlebosclerosis of the portal venous branches was less common in extrahepatic portal venous obstruction [52].
From a purely histomorphologic standpoint, the “portal tract vasculopathy” scheme by Krazinskas et al brings an insight on the pathogenesis of INCPH/PSVD. The authors reported portal vascular abnormalities similar to the ones encountered in INCPH in 88% of native and allograft liver biopsies in the absence of PH and without significant differences between the two groups [7]. The authors proposed the term “portal tract vasculopathy” and suggested that portal inflammation might be a potential etiologic factor. In the native liver group, portal tract vasculopathy was associated with hepatitis C virus (HCV) infection and increasing fibrosis whereas in the allograft group, it correlated with the severity of a synchronous acute cellular rejection episode, the presence of previous rejection episodes and increased time following transplantation [7]. Similar portal changes were described in a Japanese HCV patient without PH or cirrhosis in a three-dimensional histologic reconstruction study [84]. These studies suggest that portal vascular changes that can be seen in INCPH/PSVD may be at least in part related to portal inflammation. However, INCPH/PSVD biopsies usually do not demonstrate overt portal inflammation.
In addition, previous studies and reports have linked INCPH/PSVD to various hematologic, immunologic, infectious and genetic disease processes [1, 3, 10]. Multiple factors may coexist and contribute to the pathophysiology of this disease.
Immune dysregulation
Multiple studies have reported an association between INCPH/PSVD and disorders of immune function including congenital [85] (in particular common variable immunodeficiency (CVID)) and acquired immunodeficiencies (human immunodeficiency virus (HIV)-related) as well as autoimmune diseases [2, 10]. For example, INCPH has been described in patients with inflammatory bowel diseases, rheumatoid arthritis, systemic lupus erythematosus, autoimmune hepatitis, scleroderma, Felty’s syndrome and celiac disease among others [10, 24, 28, 29, 40, 63-65, 86]. Seropositivity for antinuclear, antimicrosomal and anti-DNA antibodies has also been reported [3, 23]. The underlying mechanism of immune-induced injury to the sinusoidal microvasculature is unclear but it appears to be related to hyperactivation of intrasinusoidal T lymphocytes [26, 35].
Hematologic factors
Prothrombotic conditions and hypercoagulable states are frequently encountered in patients diagnosed with INCPH/PSVD [2, 10, 21, 34]. Some of the histologic features of INCPH, in particular OPV, may be related to previous or recurrent thromboembolic events [8]. Likewise, INCPH/PSVD is frequently complicated by portal vein thrombosis [2, 30, 31].
In fact, INCPH/PSVD and chronic portal vein thrombosis share common etiogenic grounds [87] and they display overlapping histologic features in liver biopsies [9]. Therefore, it would be very difficult or nearly impossible to distinguish these two, or determine whether portal vein thrombosis in INCPH/PSVD represents an eventual end result of the disease or it develops de novo in a completely independent manner [73].
Infectious etiologies
INCPH/PSVD has been reported to correlate with recurrent intraabdominal infections especially in Eastern countries, although this association remains inconclusive [1, 2]. HIV may also cause a direct virus-induced damage to the sinusoidal endothelial cells [88]. Endothelial cell injury may also occur as a result of prolonged exposure to antiretroviral treatment [31]. Additionally, HIV-related acquired protein S deficiency induces a state of hypercoagulability, a known risk factor for INCPH/PSVD [89]. Given the multitude of abnormalities seen in this population, the exact relationship between HIV and INCPH/PSVD appears to remain speculative [10].
Hereditary predispositions
Familial clusters of INCPH/PSVD have been reported, many of which were linked to a specific HLA haplotype (HLA-DR3) [36]. In familial cases, mutations in KCNN3 and DGUOK genes have been implicated. These alterations are transmitted in an autosomal dominant and recessive fashion, respectively [90, 91]. Furthermore, those with certain congenital syndromes and hereditary diseases are at increased risk of developing INCPH/PSVD (i.e., Turner syndrome, Adams-Oliver syndrome, phosphomannose isomerase deficiency, cystic fibrosis) [2, 46, 92, 93]. Lastly, HIV patients who harbor specific single nucleotide polymorphisms (SNPs) in genes encoding the enzymes involved in purine metabolism are at increased risk of INCPH/PSVD when exposed to didanosine, an anti-retroviral agent [94].
Drug exposure
Exposure to variable drugs such as azathioprine, oxaliplatin, stavudine, didanosine and thioguanine has been linked to the development of INCPH/PSVD [2, 10].
Idiopathic
Some INCPH/PSVD patients present without any known risk factors. Cazals-Hatem et al reported that no risk factors of INCPH were identified in 57% of patients with histologic OPV with or without PH at presentation [40].
Metabolic factors
Given the broadened definitional spectrum of PSVD, liver diseases that can cause pre-cirrhotic PH that are not included in the “to be excluded” conditions of PSVD would meet the diagnostic criteria of PSVD. The prime examples would be alcoholic fatty liver disease and NAFLD.
There is growing evidence that PH can occur in alcoholic fatty liver disease and NAFLD even in the absence of significant fibrosis/cirrhosis and that it correlates with the degree of steatosis [95-101]. Although the exact etiopathogenesis remains unclear, multiple hypotheses have been proposed. Enlarged and ballooned fatty hepatocytes impair portal blood flow by narrowing the sinusoidal space. Consequent sinusoidal endothelial cell injury and loss of fenestrations facilitate entrapment of blood cells with increased deposition of extracellular matrix in the space of Disse. Also, the endothelial cells’ inhibitory control on hepatic stellate cells diminishes leading to unrestrained vasoconstriction, further impeding the sinusoidal blood flow. Resultant tissue hypoxia promotes inflammation, fibrosis and neovascularization by triggering Kupffer cell activation [95].
Animal studies have also shown that steatohepatitis induces PH in the absence of fibrosis as a result of a hyperdynamic splanchnic circulation with increased portal blood flow and impaired arteriolar response to vasoconstrictors [97, 102]. Similarly, Mendes et al found that obesity and diabetes are independently associated with the development of PH [102]. In alcoholic liver disease, central sclerosing hyaline necrosis is believed to be the contributory factor to the development of pre-cirrhotic PH [101].
Likewise, Zuo et al found that individual morphologic INCPH/PSVD lesions were significantly more common in steatotic liver specimens when compared to incidental biopsies, in the absence of PH [13]. This further corroborates the fact that fatty liver disease may contribute to the vascular remodeling seen in INCPH/PSVD though it will be difficult to determine which component is the main driver accountable for the histologic changes (Fig. 3).
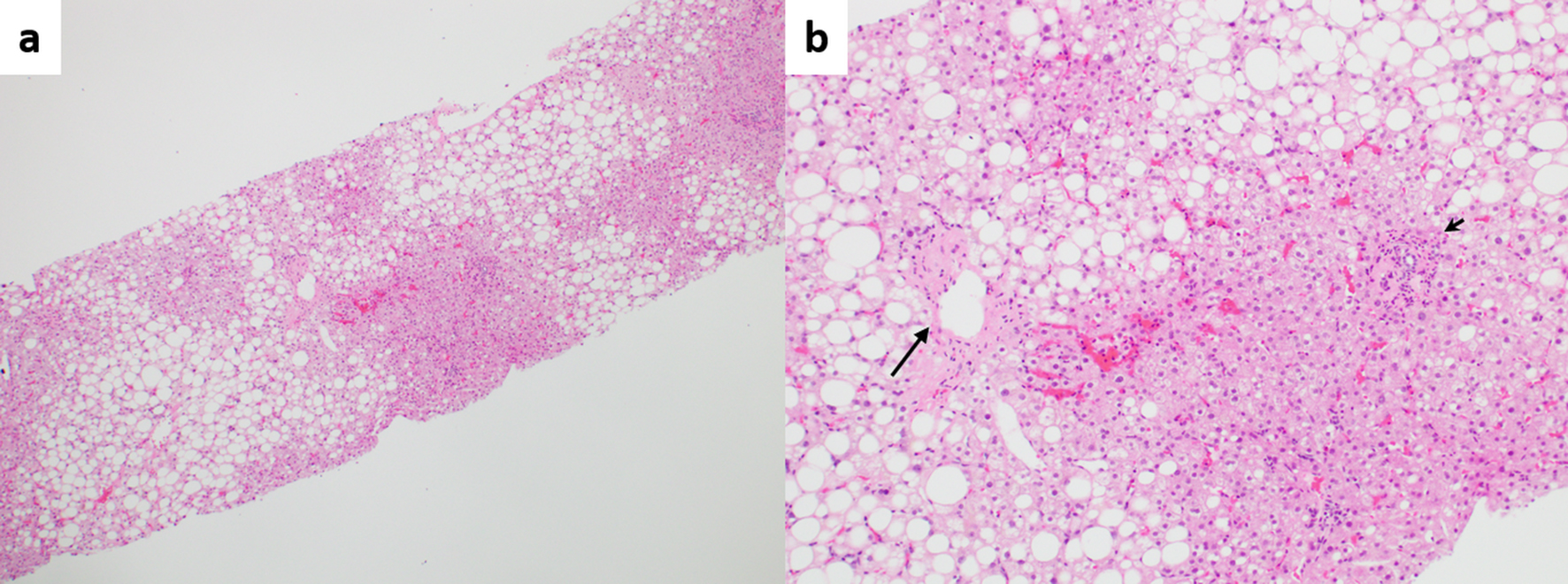 Click for large image | Figure 3. (a) Fatty liver disease without cirrhosis or advanced fibrosis, but with portal hypertension. Zonal steatosis is noted (hematoxylin and eosin (H&E), × 40). (b) Higher magnification view showing pericentral fibrosis (long arrow) probably secondary to fatty liver disease, and phlebosclerosis (obliterative portal venopathy, short arrow), possibly secondary to concurrent porto-sinusoidal vascular disease (H&E, × 100). |
Etiopathogenic associations with PSVD are summarized in Figure 4.
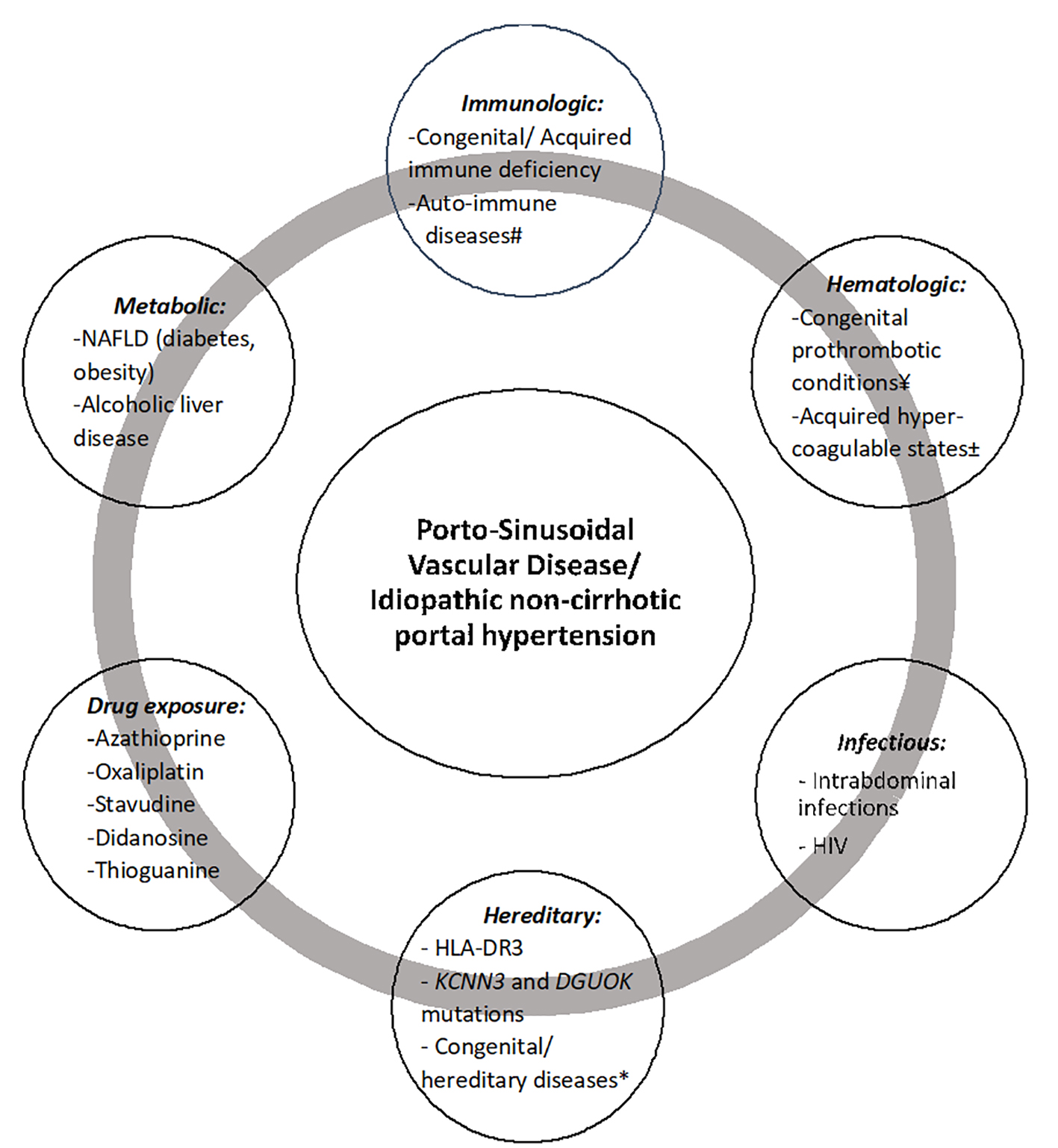 Click for large image | Figure 4. Etiopathogenic associations with porto-sinusoidal vascular disease. #Systemic lupus erythematosus, rheumatoid arthritis, scleroderma, Sjogren’s syndrome, inflammatory bowel diseases, celiac disease, autoimmune hepatitis, Felty’s syndrome. ¥Protein C or S deficiency, factor V Leiden, factor II mutation, antithrombin III deficiency. ±Antiphospholipid syndrome, malignancy, ADAMTS 13 deficiency, oral contraceptive use. *Turner syndrome, Adams-Oliver syndrome, cystic fibrosis, phosphomannose isomerase deficiency. NAFLD: non-alcoholic fatty liver disease; HIV: human immunodeficiency virus. |
| Diagnosis | ▴Top |
The diagnosis of INCPH/PSVD relies on a constellation of clinical, radiologic and histologic findings. Liver biopsy is indispensable for documenting the absence of cirrhosis and as previously stated, can establish the diagnosis by itself when specific morphologic lesions of PSVD are identified [2].
Clinical presentation
Patients usually present with signs and symptoms of PH to include gastroesophageal (present in up to two-thirds of patients at the time of diagnosis) and anorectal varices, hypersplenism and thrombocytopenia [2, 80, 103]. Gastrointestinal bleeding secondary to ruptured varices is the most frequently encountered presenting symptom seen in up to 50% of patients [80]. There are 20-50% of patients presenting with ascites which often occurs in the setting of concurrent gastrointestinal infection or hemorrhage. Some patients, however, may be completely asymptomatic and signs of PH are discovered incidentally either by radiologic or endoscopic means [34]. Less commonly, hepatic encephalopathy, hepatorenal and hepatopulmonary syndrome may develop [2, 80]. Hepatic synthetic function is relatively preserved, but progression to advanced hepatic insufficiency requiring liver transplantation occurs in 4-19% of cases [40, 47, 104].
Subsequent portal vein thrombosis is frequently seen in INCPH/PSVD patients. In a study by Matsutani et al, nine (41%) of 22 patients with INCPH developed portal vein thrombosis after 12 years of follow-up, at a significantly higher incidence compared to cirrhotic patients. Also, the development of portal vein thrombosis was an indicator of worse prognosis [30]. Similarly, portal vein thrombosis developed in 75% of INCPH patients with concurrent HIV infection after a median follow-up of 15 months [31].
As stated previously, clinical and or radiologic evidence of PH is not a prerequisite for the diagnosis of PSVD. As such, PSVD may be found during the workup of chronic abnormal liver function tests of unknown etiology. Elevated serum levels of transaminases, alkaline phosphatase, gamma glutamyl transferase and bilirubin have been variably reported. Not uncommonly, patients may be referred to hematologists for thrombocytopenia secondary to hypersplenism or abnormal coagulation tests due to advanced liver dysfunction [79, 105]. However, PSVD may also be an incidental finding. For example, in a study by Lee et al, histologic features of INCPH were identified in all liver biopsies from 13 dialysis patients but only a third (four of 13) had underlying PH, qualifying for INCPH [17]. Among the remaining nine patients without PH, six (66%) would have met the diagnostic criteria for PSVD given the presence of OPV in the liver specimens. Regardless, the authors speculated that the findings of histologic features of INCPH were reflective of the presence of known INCPH/PSVD risk factors that are commonly encountered in this patient population. PSVD without PH was also reported as a form of vascular remodeling in regressed fibrosis following phlebotomy in hemochromatosis patients [14]. On the other hand, some PSVD patients without PH at presentation may develop overt PH on follow-up studies [40, 79]. In Cazals-Hatem et al’s study, six (40%) of 15 patients with OPV in liver specimens at presentation but without PH or portal vein thrombosis developed PH after a mean follow-up of 8.6 years [40].
Radiologic features
Imaging studies can be used to document the presence of PH either indirectly by noninvasive means (i.e., splenomegaly, porto-systemic collaterals) or directly by measuring the hepatic venous pressures via catheterization of the hepatic veins. They can also identify disease processes that should be otherwise excluded in order to diagnose PSVD (i.e., schistosomiasis, congenital hepatic fibrosis). Although radiologic findings often lack sufficient specificity to establish a diagnosis, there are some features that favor INCPH/PSVD.
First, combined hypertrophy of the caudate lobe and segment IV of the liver is suggestive of INCPH/PSVD whereas cirrhosis is usually associated with segment IV atrophy [41, 44]. Thickened hyperechoic walls of intrahepatic portal branches on ultrasound and increased periportal signal intensity on T2-weighted MRI can be seen in some INCPH/PSVD patients and may represent periportal fibrosis [48, 53]. In addition, a normal or mildly elevated HVPG in the setting of overt stigmata of PH would support the diagnosis of INCPH/PSVD as it is indicative of a presinusoidal form of PH [32]. One caveat is that the presence of intrahepatic vein-to-vein communications, commonly seen in INCPH/PSVD, precludes an adequate assessment of HVPG and subsequently would underestimate the actual value of the portal venous pressure [32, 106]. Also, it is important to note that even though preserved liver volume and the absence of nodular contours would favor INCPH/PSVD in the early stages of the disease [48], hepatic surface nodularity resembling cirrhosis can be seen in INCPH/PSVD [10, 55] (Fig. 5). Abnormalities of the intrahepatic portal venous system (i.e., reduced caliber, diminished visibility and occlusive thrombosis) are more commonly seen in INCPH/PSVD than in cirrhosis [41].
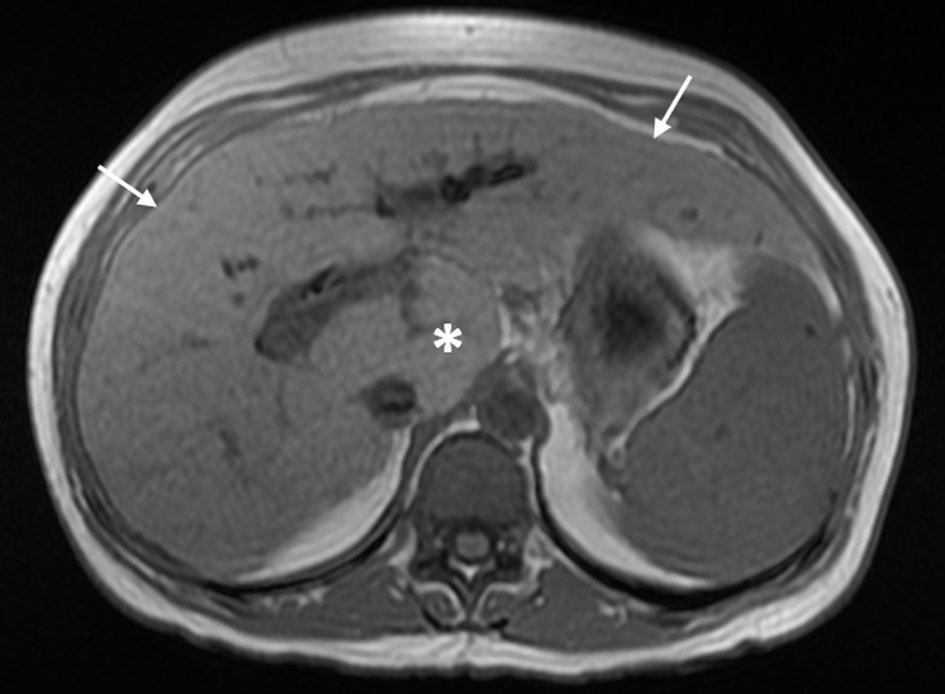 Click for large image | Figure 5. Magnetic resonance imaging with contrast shows mild nodular contour of the liver surface (arrows) and relative hypertrophy of the caudate lobe (*) in porto-sinusoidal vascular disease. |
Data on liver stiffness measurement (via transient elastography) are limited in INCPH/PSVD. However, recent studies have shown that it can be useful in distinguishing INCPH/PSVD from cirrhosis [32, 33, 107]. INCPH/PSVD has significantly lower mean liver stiffness compared to that of cirrhosis. In Elkrief et al’s study, the cutoff values of 10 and 20 kPa were found to have sufficient sensitivity and specificity, respectively, in distinguishing the two [107]. For example, liver stiffness values lower than 10 kPa were highly suggestive of INCPH/PSVD in patients with signs of PH whereas levels exceeding 20 kPa effectively excluded the disease [107]. Additionally, hepatic venography shows frequent veno-venous shunts in INCPH/PSVD with narrower angles between the large veins and their branches when compared to cirrhosis [108].
Histomorphologic findings
INCPH/PSVD lesions can be observed both in the portal tracts and within hepatic lobules. The definition of PSVD identifies three specific histologic lesions that are sufficient, per se, to establish the diagnosis even in the absence of clinical or radiologic features of PH: OPV, nodular regenerative hyperplasia and incomplete septal fibrosis/cirrhosis [2] (Table 2).
OPV, also known as phlebosclerosis [38], is considered to be the hallmark of PSVD. It is believed to be the initiating event leading to increased portal flow resistance and sustained presinusoidal hypertension [79]. OPV is characterized by mural fibrosis and thickening of intrahepatic small and medium-sized portal vein branches with subsequent luminal narrowing, obliteration and venopenia [12, 42]. However, it is important to distinguish these features from portal dyads, a normal histologic variant, where the lack of portal veins may be misinterpreted as OPV [13].
Nodular regenerative hyperplasia was first introduced by Steinert as a liver parenchymal micronodularity in the absence of fibrosis [60]. It appears to be the result of OPV with subsequent uneven perfusion of the liver parenchyma. The nodules are usually small (1 - 3 mm), ill-defined and composed of the hyperplastic central zones delineated by peripherally compressed atrophic hepatic cell plates. The nodular architecture is better appreciated with a reticulin stain that highlights the condensed reticulin network at the periphery of the nodules [61, 79]. Cytokeratin 7 immunostain may also be helpful as it is often expressed in atrophic hepatocytes [109].
Incomplete septal fibrosis/cirrhosis is characterized by the presence of thin fibrous septae emanating from a portal tract and ending blindly within the hepatic lobule [79]. Although a vague and poorly demarcated hepatic nodularity can be seen, complete cirrhotic-type nodules should not be seen in incomplete septal fibrosis/cirrhosis (Fig. 6).
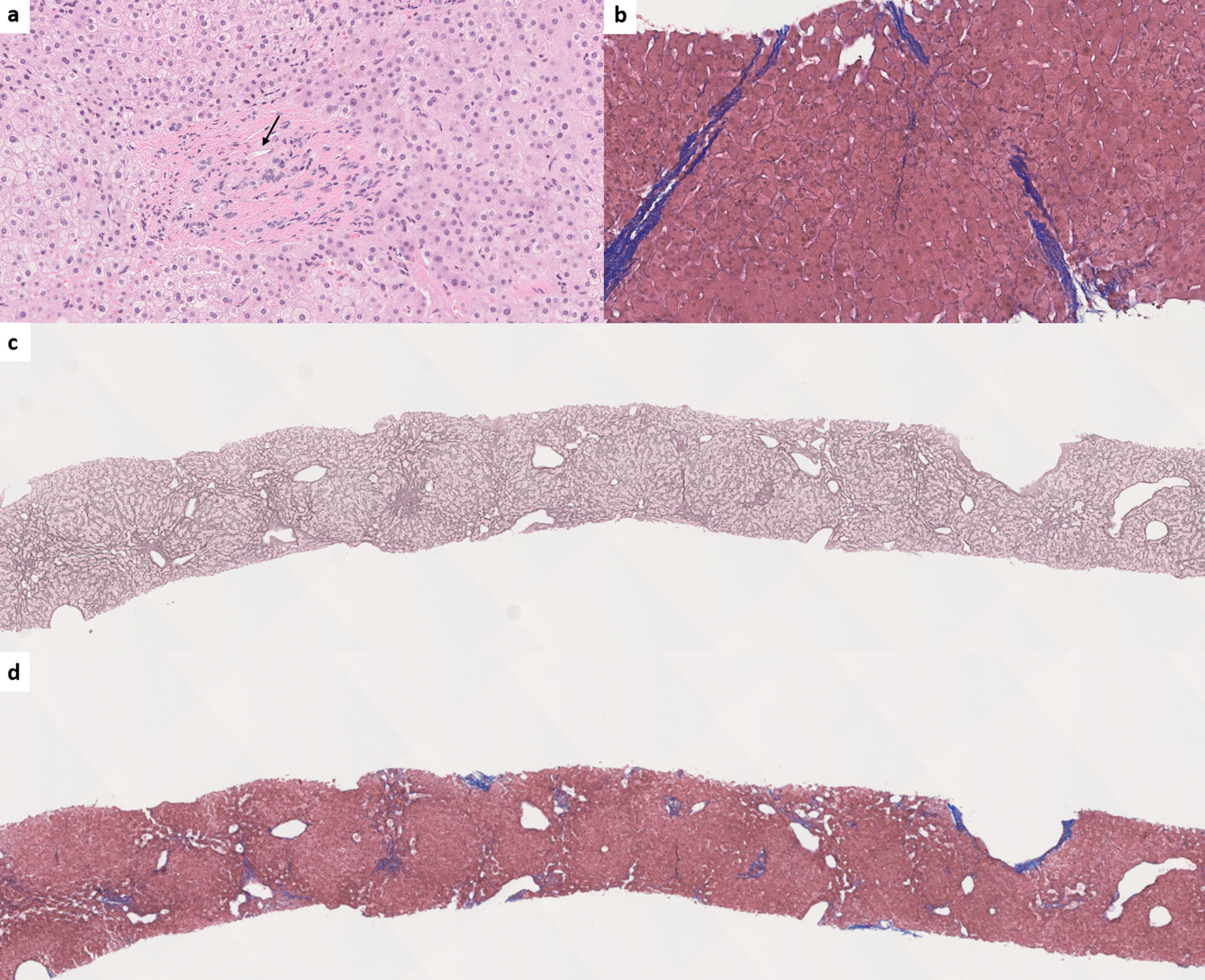 Click for large image | Figure 6. (a) Obliterative portal venopathy (also known as phlebosclerosis) with inconspicuous portal venous branch (arrow) (hematoxylin and eosin, × 200). (b) Incomplete septal fibrosis or cirrhosis (Masson-trichrome, × 150). (c) Nodular regenerative hyperplasia. The nodularity is highlighted by reticulin special stain (reticulin, × 30). (d) Trichrome stain shows the absence of cirrhosis in nodular regenerative hyperplasia (Masson-trichrome, × 30). |
PSVD scheme defines not specific histologic lesions as those that can be associated with INCPH/PSVD but lack specificity. These include portal tract abnormalities such as aberrant vessels (i.e., periportal shunting vessels, herniated portal veins branches, multiplicity of portal vessels within a single portal tract, dilatation of arteries), irregular distribution of the portal tracts and central veins, non-zonal sinusoidal dilatation and mild perisinusoidal fibrosis [2] (Table 2). Additionally, portal tract remnants (defined as portal tract smaller than twice the size of a bile duct) are frequently seen in INCPH/PSVD [9, 13, 14, 18, 79].
The most common lobular abnormality seen in INCPH/PSVD is sinusoidal dilatation [1, 12, 40, 42, 79] as INCPH/PSVD is a form of vascular pattern injury. However, sinusoidal dilatation is not specific and can be associated with other vascular, neoplastic, inflammatory, infectious and medication-related etiologies [77]. It is important to note that in INCPH/PSVD, sinusoidal dilatation is random and non-zonal [2], whereas it is predominantly centrilobular when associated with usual etiogenic factors and mainly periportal when associated with oral contraceptive use [71]. Interestingly, a recent reproducibility study on individual histologic features of INCPH/PSVD has shown that the recognition of sinusoidal dilatation was relatively reproducible amongst nine experienced liver pathologists, although the zone in which sinusoidal dilatation was identified was not specified. This indicates that recognizing sinusoidal dilatation may be a helpful clue in considering the diagnosis of INCPH/PSVD in the right clinical context [18] (Fig. 7).
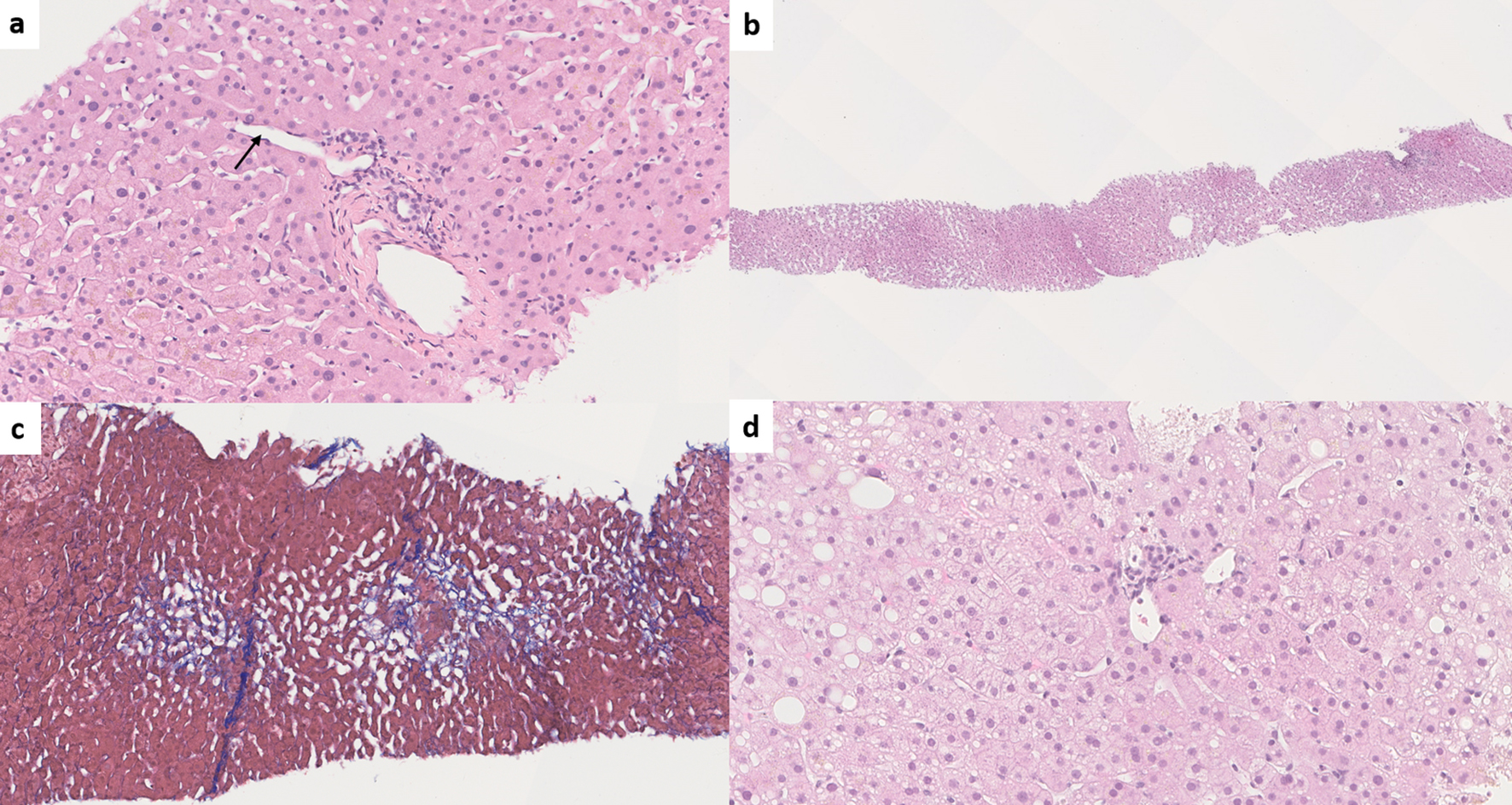 Click for large image | Figure 7. (a) Paraportal shunt vessel (arrow) in idiopathic non-cirrhotic portal hypertension/porto-sinusoidal vascular disease (INCPH/PSVD) (hematoxylin and eosin (H&E), × 200). (b) Irregularly distributed portal tracts and central veins in INCPH/PSVD. Non-zonal sinusoidal dilatation is also noted (H&E, × 50). (c) Mild perisinusoidal fibrosis (Masson-trichrome, × 130). (d) Rudimentary portal tract in INCPH/PSVD (H&E, × 250). |
In an attempt to standardize the variable terminologies used for these histologic lesions, specifically for INCPH, the International Liver Pathology Study Group proposed “portal vein stenosis” for phlebosclerosis and OPV, “herniated portal vein” for herniated portal vein branches, “hypervascularized portal tract” for multiplicity of portal vessels and “periportal abnormal vessels” for periportal shunting vessels [11].
Interobserver variability
Histologic lesions of INCPH/PSVD are heterogenous and frequently overlap with other entities thus, display low interobserver agreement even among experienced liver pathologists [18, 66, 110]. Liang et al recently have shown that the interobserver agreement can be improved when OPV is recognized as the sole independent predictor of INCPH/PSVD. Further, the authors proposed three different categories for OPV: consistent with OPV, indeterminate for OPV and not consistent with OPV [110]. Whether this novel categorization improves diagnostic accuracy of INCPH/PSVD is yet to be determined and needs to be validated by future studies.
Inflow vs. outflow vascular abnormality
INCPH/PSVD is a presinusoidal form of PH thus falls within the spectrum of inflow vascular abnormality. Another cardinal example of inflow vascular abnormality with similar clinical and morphologic pictures is chronic non-cirrhotic, non-malignant portal vein thrombosis [73]. Therefore, despite overlapping clinical and morphologic features, INCPH/PSVD should be distinguished from hepatic venous outflow abnormality. Histologic hallmark of INCPH/PSVD is OPV in the portal tracts and other vascular and parenchymal changes are considered secondary to OPV [2]. In contrast, in outflow vascular abnormality (as in BCS, SOS and congestive hepatopathy), the morphologic changes predominate in the central zones (congestion, necrosis and eventual fibrosis) and affect the central veins [73, 105]. Additionally, despite similar presenting symptoms, the clinicoradiologic context would be quite different between inflow and outflow abnormality. For example, the presence of heart failure in congestive hepatopathy, history of hematopoietic stem cell transplantation and drug exposure in SOS and radiologic evidence of hepatic venous outflow obstruction in BCS would essentially preclude the diagnosis of INCPH/PSVD. However, in a limited sample or advanced diseases, determining primary lesion (portal tract vs. lobule) can be challenging.
Diagram summarizing the diagnostic approach for INCPH and PSVD is illustrated in Figure 8.
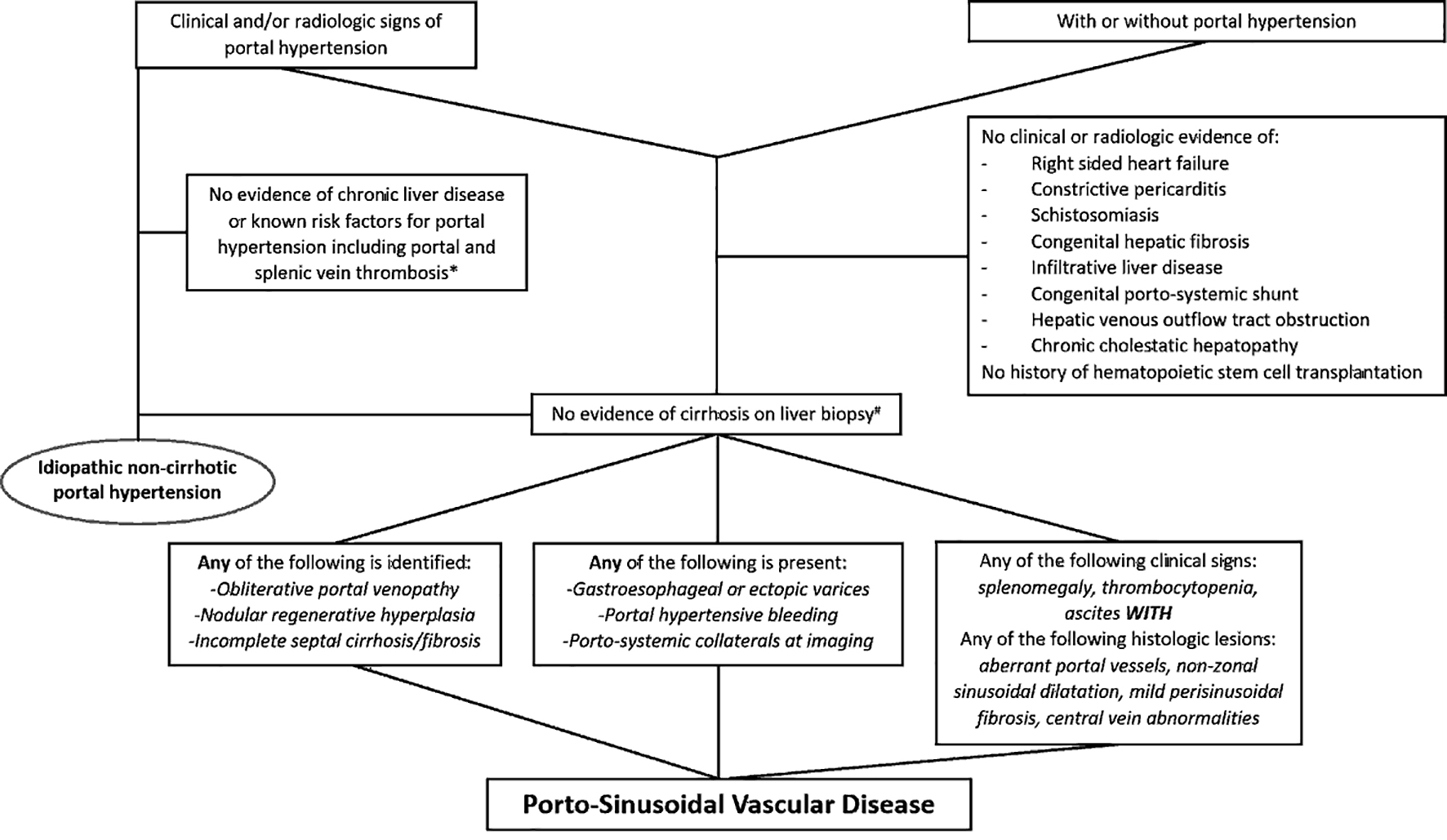 Click for large image | Figure 8. Diagram summarizing the diagnostic approach for idiopathic non-cirrhotic portal hypertension and porto-sinusoidal vascular disease. *Portal vein thrombosis may be seen along the natural disease course of idiopathic non-cirrhotic portal hypertension. #Biopsy adequacy criteria are available for porto-sinusoidal vascular disease (core liver biopsy ≥ 20 mm in length or featuring ≥ 10 portal tracts, or when considered adequate by an expert pathologist). |
| Management | ▴Top |
There is no specific treatment for PSVD, especially in patients without PH as some of these patients may probably never develop overt PH. In fact, the natural history of PSVD in the absence of PH is largely unknown [2] and large-scale prospective studies are needed in order to implement management recommendations in this patient population. Currently, the initial management for patients with INCPH and PSVD with PH is to identify and treat the underlying conditions known to be associated with the disease [10].
In INCPH, complications related to PH are managed in a similar fashion with those in patients with cirrhotic PH [10, 80]. These patients should be screened regularly for gastroesophageal varices and prophylactic measures for variceal bleeding should be implemented when varices are identified. Regarding the timing of endoscopic follow-up and the type of prophylaxis recommended in INCPH, the few studies available failed to draw solid conclusions as only a small number of patients were studied [111, 112]. Therefore, in the absence of specific and universally implemented guidelines, the screening frequency and primary and secondary prophylaxis for variceal bleeding in INCPH are similar to those recommended for cirrhotic PH. The prophylactic measures include the use of non-specific beta-adrenergic blockage and endoscopic band ligation [2, 56, 80, 113]. Similarly, the management for ascites and hepatic encephalopathy is identical to those in cirrhotic patients. Transjugular intrahepatic porto-systemic shunt (TIPS) is reserved for patients with severe variceal bleeding and refractory ascites who fail conservative treatment. An international multicenter study showed that the outcomes of TIPS were favorable with a 2-year survival rate of 80% in INCPH patients [114].
Treatment with anticoagulants is often considered in INCPH given the frequent association with portal vein thrombosis and underlying hypercoagulable state. However, routine use of anticoagulants in INCPH patients is currently not recommended due to insufficient evidence of benefit [113, 115]. On the other hand, current guidelines recommend biannual Doppler ultrasound in INCPH/PSVD patients for early detection of portal vein thrombosis [115]. Liver transplantation is a valid option for INCPH patients with advanced hepatocellular insufficiency. The data regarding outcomes after liver transplantation in INCPH patients are limited, but appear overall favorable [6, 62].
| Clinical Outcomes | ▴Top |
In general, INCPH/PSVD patients fare considerably better than those with cirrhosis. Patients with INCPH/PSVD are less likely to develop ascites or hepatic encephalopathy than cirrhosis patients. Even though variceal bleeding is more common, the associated mortality is significantly lower compared to cirrhosis patients [34, 54]. This might be due to relatively preserved hepatocellular function in INCPH/PSVD [10].
Recent study has shown that the clinical course of PSVD without PH is more favorable than that of INCPH/PSVD with PH. Woran et al compared PSVD patients with PH (INCPH) to PSVD patients without PH and found the former to have higher Child-Pughes scores, higher prevalence of liver decompensation and higher liver-related mortality compared to the latter [116].
Progression toward advanced hepatic failure and the development of hepatocellular carcinoma are unusual in INCPH/PSVD [104, 117, 118]. Hence, routine ultrasonographic screening for hepatocellular carcinoma is generally not recommended in INCPH/PSVD patients [80].
| Summary | ▴Top |
In summary, INCPH/PSVD remains relatively under-recognized and its incidence is probably underestimated. INCPH can be diagnosed when histologic features of INCPH/PSVD are identified in a non-cirrhotic liver biopsy from patients with PH of unknown etiology. Minimum histologic diagnostic criteria are not defined in INCPH. On the other hand, PSVD may be diagnosed in the absence of PH when aforementioned specific histologic features are present on liver biopsy. When not specific histologic features are only present, certain clinical signs of PH are required to establish a diagnosis of PSVD. Future studies and investigations are needed in order to better define the significance of subclinical PSVD and to standardize diagnostic and therapeutic strategies. Although we acknowledge that PSVD is a broader term, given the importance of PH in patients’ management, the term INCPH might be a better choice than PSVD with PH. The term PSVD without PH would be a reasonable histologic diagnosis that can be used to accrue relevant clinical data.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Author Contributions
The authors made substantial contributions to the conception of the work (XL, HL), drafting the work (MK) and revised it critically for important intellectual content (XL, SB, HL). The authors approve final version that is to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
INCPH: idiopathic non-cirrhotic portal hypertension; PH: portal hypertension; PSVD: porto-sinusoidal vascular disease; HVPG: hepatic venous pressure gradient; NCPH: non-cirrhotic portal hypertension; OPV: obliterative portal venopathy; VALDIG: Vascular Liver Disease Interest Group; BCS: Budd-Chiari syndrome; HVOTO: hepatic venous outflow tract obstruction; MRI: magnetic resonance imaging; MELD: model for end-stage liver disease; SOS: sinusoidal obstruction syndrome; VOD: veno-occlusive disease; HSCT: hematopoietic stem cell transplantation; NOSD: non-obstructive sinusoidal dilatation; PSOB: post-sinusoidal outflow block; CPS: congenital porto-systemic shunt; HCV: hepatitis C virus; CVID: common variable immunodeficiency; HIV: human immunodeficiency virus; NAFLD: nonalcoholic fatty liver disease; TIPS: transjugular intrahepatic porto-systemic shunt
| References | ▴Top |
- Schouten JN, Garcia-Pagan JC, Valla DC, Janssen HL. Idiopathic noncirrhotic portal hypertension. Hepatology. 2011;54(3):1071-1081.
doi pubmed - De Gottardi A, Rautou PE, Schouten J, Rubbia-Brandt L, Leebeek F, Trebicka J, Murad SD, et al. Porto-sinusoidal vascular disease: proposal and description of a novel entity. Lancet Gastroenterol Hepatol. 2019;4(5):399-411.
doi - Rajekar H, Vasishta RK, Chawla YK, Dhiman RK. Noncirrhotic portal hypertension. J Clin Exp Hepatol. 2011;1(2):94-108.
doi - CDC. Global Health, Division of Parasitic Diseases and Malaria- Schistosomiasis. https://www.cdc.gov/parasites/schistosomiasis/epi.html. February, 2020.
- Tetangco EP, Silva RG, Lerma EV. Portal hypertension: Etiology, evaluation, and management. Dis Mon. 2016;62(12):411-426.
doi pubmed - Krasinskas AM, Eghtesad B, Kamath PS, Demetris AJ, Abraham SC. Liver transplantation for severe intrahepatic noncirrhotic portal hypertension. Liver Transpl. 2005;11(6):627-634; discussion 610-621.
doi pubmed - Krasinskas AM, Goldsmith JD, Burke A, Furth EE. Abnormal intrahepatic portal vasculature in native and allograft liver biopsies: a comparative analysis. Am J Surg Pathol. 2005;29(10):1382-1388.
doi pubmed - Schouten JN, Verheij J, Seijo S. Idiopathic non-cirrhotic portal hypertension: a review. Orphanet J Rare Dis. 2015;10:67.
doi pubmed - Verheij J, Schouten JN, Komuta M, Nevens F, Hansen BE, Janssen HL, Roskams T. Histological features in western patients with idiopathic non-cirrhotic portal hypertension. Histopathology. 2013;62(7):1083-1091.
doi pubmed - Lee H, Rehman AU, Fiel MI. Idiopathic Noncirrhotic Portal Hypertension: An Appraisal. J Pathol Transl Med. 2016;50(1):17-25.
doi pubmed - Guido M, Alves VAF, Balabaud C, Bathal PS, Bioulac-Sage P, Colombari R, Crawford JM, et al. Histology of portal vascular changes associated with idiopathic non-cirrhotic portal hypertension: nomenclature and definition. Histopathology. 2019;74(2):219-226.
doi pubmed - Guido M, Sarcognato S, Sonzogni A, Luca MG, Senzolo M, Fagiuoli S, Ferrarese A, et al. Obliterative portal venopathy without portal hypertension: an underestimated condition. Liver Int. 2016;36(3):454-460.
doi pubmed - Zuo C, Chumbalkar V, Ells PF, Bonville DJ, Lee H. Prevalence of histological features of idiopathic noncirrhotic portal hypertension in general population: a retrospective study of incidental liver biopsies. Hepatol Int. 2017;11(5):452-460.
doi pubmed - El Jabbour T, McHugh KE, Patil DT, Zuo C, Koo BH, Kim S, Lee H. Histologic lesions of Porto-Sinusoidal vascular disease following phlebotomy in hemochromatosis. Gastroenterology Res. 2020;13(1):32-39.
doi pubmed - Wanless IR, Bernier V, Seger M. Intrahepatic portal vein sclerosis in patients without a history of liver disease. An autopsy study. Am J Pathol. 1982;106(1):63-70.
- Nakanuma Y, Hoso M, Sasaki M, Terada T, Katayanagi K, Nonomura A, Kurumaya H, et al. Histopathology of the liver in non-cirrhotic portal hypertension of unknown aetiology. Histopathology. 1996;28(3):195-204.
doi pubmed - Lee H, Ainechi S, Singh M, Ells PF, Sheehan CE, Lin J. Histological spectrum of idiopathic noncirrhotic portal hypertension in liver biopsies from dialysis patients. Int J Surg Pathol. 2015;23(6):439-446.
doi pubmed - Kmeid M, Zuo C, Lagana SM, Choi WT, Lin J, Yang Z, Liu X, et al. Interobserver study on histologic features of idiopathic non-cirrhotic portal hypertension. Diagn Pathol. 2020;15(1):129.
doi pubmed - Kobayashi Y, Inokuchi K, Saku M. Epidemiology of idiopathic portal hypertension based on a nation-wide survey. In: Suguira M, ed. Report of the Ministry of Health and Welfare Research Committee on Idiopathic Portal Hypertension. Tokyo: Japan Ministry of Health and Welfare. 1976:10-15.
- Mukta V, Panicker LC, Sivamani K, Goel A, Basu D, Dhanapathi H. Non-cirrhotic portal fibrosis at a tertiary care centre in South India. Trop Doct. 2017;47(1):26-30.
doi pubmed - Hillaire S, Bonte E, Denninger MH, Casadevall N, Cadranel JF, Lebrec D, Valla D, et al. Idiopathic non-cirrhotic intrahepatic portal hypertension in the West: a re-evaluation in 28 patients. Gut. 2002;51(2):275-280.
doi pubmed - Okuda K, Nakashima T, Okudaira M, Kage M, Aida Y, Omata M, Sugiura M, et al. Liver pathology of idiopathic portal hypertension. Comparison with non-cirrhotic portal fibrosis of India. The Japan idiopathic portal hypertension study. Liver. 1982;2(3):176-192.
doi pubmed - Nakanuma Y, Nonomura A, Hayashi M, Doishita K, Takayanagi N, Uchida T, Obata Y, et al. Pathology of the liver in "idiopathic portal hypertension" associated with autoimmune disease. The Ministry of Health and Welfare Disorders of Portal Circulation Research Committee. Acta Pathol Jpn. 1989;39(9):586-592.
doi - Saito K, Nakanuma Y, Takegoshi K, Ohta G, Obata Y, Okuda K, Kameda H. Non-specific immunological abnormalities and association of autoimmune diseases in idiopathic portal hypertension. A study by questionnaire. Hepatogastroenterology. 1993;40(2):163-166.
- Oikawa H, Masuda T, Sato S, Yashima A, Suzuki K, Sato S, Satodate R. Changes in lymph vessels and portal veins in the portal tract of patients with idiopathic portal hypertension: a morphometric study. Hepatology. 1998;27(6):1607-1610.
doi pubmed - Yamaguchi N, Tokushige K, Haruta I, Yamauchi K, Hayashi N. Analysis of adhesion molecules in patients with idiopathic portal hypertension. J Gastroenterol Hepatol. 1999;14(4):364-369.
doi pubmed - Okudaira M, Ohbu M, Okuda K. Idiopathic portal hypertension and its pathology. Semin Liver Dis. 2002;22(1):59-72.
doi pubmed - Tsuneyama K, Harada K, Katayanagi K, Watanabe K, Kurumaya H, Minato H, Nakanuma Y. Overlap of idiopathic portal hypertension and scleroderma: report of two autopsy cases and a review of literature. J Gastroenterol Hepatol. 2002;17(2):217-223.
doi pubmed - Kogawa H, Migita K, Ito M, Takii Y, Daikoku M, Nakao M, Miyashita T, et al. Idiopathic portal hypertension associated with systemic sclerosis and Sjogren's syndrome. Clin Rheumatol. 2005;24(5):544-547.
doi pubmed - Matsutani S, Maruyama H, Akiike T, Kobayashi S, Yoshizumi H, Okugawa H, Fukuzawa T, et al. Study of portal vein thrombosis in patients with idiopathic portal hypertension in Japan. Liver Int. 2005;25(5):978-983.
doi pubmed - Chang PE, Miquel R, Blanco JL, Laguno M, Bruguera M, Abraldes JG, Bosch J, et al. Idiopathic portal hypertension in patients with HIV infection treated with highly active antiretroviral therapy. Am J Gastroenterol. 2009;104(7):1707-1714.
doi pubmed - Seijo S, Reverter E, Miquel R, Berzigotti A, Abraldes JG, Bosch J, Garcia-Pagan JC. Role of hepatic vein catheterisation and transient elastography in the diagnosis of idiopathic portal hypertension. Dig Liver Dis. 2012;44(10):855-860.
doi pubmed - Furuichi Y, Moriyasu F, Taira J, Sugimoto K, Sano T, Ichimura S, Miyata Y, et al. Noninvasive diagnostic method for idiopathic portal hypertension based on measurements of liver and spleen stiffness by ARFI elastography. J Gastroenterol. 2013;48(9):1061-1068.
doi pubmed - Siramolpiwat S, Seijo S, Miquel R, Berzigotti A, Garcia-Criado A, Darnell A, Turon F, et al. Idiopathic portal hypertension: natural history and long-term outcome. Hepatology. 2014;59(6):2276-2285.
doi pubmed - Kotani K, Kawabe J, Morikawa H, Akahoshi T, Hashizume M, Shiomi S. Comprehensive screening of gene function and networks by DNA microarray analysis in Japanese patients with idiopathic portal hypertension. Mediators Inflamm. 2015;2015:349215.
doi pubmed - Sarin SK, Mehra NK, Agarwal A, Malhotra V, Anand BS, Taneja V. Familial aggregation in noncirrhotic portal fibrosis: a report of four families. Am J Gastroenterol. 1987;82(11):1130-1133.
- Sood V, Lal BB, Khanna R, Rawat D, Bihari C, Alam S. Noncirrhotic portal fibrosis in pediatric population. J Pediatr Gastroenterol Nutr. 2017;64(5):748-753.
doi pubmed - Mikkelsen WP, Edmondson HA, Peters RL, Redeker AG, Reynolds TB. Extra- and intrahepatic portal hypertension without cirrhosis (hepatoportal sclerosis). Ann Surg. 1965;162(4):602-620.
doi pubmed - Nayak NC, Ramalingaswami V. Obliterative portal venopathy of the liver. Associated with so-called idiopathic portal hypertension or tropical splenomegaly. Arch Pathol. 1969;87(4):359-369.
- Cazals-Hatem D, Hillaire S, Rudler M, Plessier A, Paradis V, Condat B, Francoz C, et al. Obliterative portal venopathy: portal hypertension is not always present at diagnosis. J Hepatol. 2011;54(3):455-461.
doi pubmed - Glatard AS, Hillaire S, d'Assignies G, Cazals-Hatem D, Plessier A, Valla DC, Vilgrain V. Obliterative portal venopathy: findings at CT imaging. Radiology. 2012;263(3):741-750.
doi pubmed - Aggarwal S, Fiel MI, Schiano TD. Obliterative portal venopathy: a clinical and histopathological review. Dig Dis Sci. 2013;58(10):2767-2776.
doi pubmed - Franchi-Abella S, Fabre M, Mselati E, De Marsillac ME, Bayari M, Pariente D, Jacquemin E, et al. Obliterative portal venopathy: a study of 48 children. J Pediatr. 2014;165(1):190-193 e192.
doi pubmed - Arora A, Sarin SK. Multimodality imaging of obliterative portal venopathy: what every radiologist should know. Br J Radiol. 2015;88(1046):20140653.
doi pubmed - Besmond C, Valla D, Hubert L, Poirier K, Grosse B, Guettier C, Bernard O, et al. Mutations in the novel gene FOPV are associated with familial autosomal dominant and non-familial obliterative portal venopathy. Liver Int. 2018;38(2):358-364.
doi pubmed - Girard M, Amiel J, Fabre M, Pariente D, Lyonnet S, Jacquemin E. Adams-Oliver syndrome and hepatoportal sclerosis: occasional association or common mechanism? Am J Med Genet A. 2005;135(2):186-189.
doi pubmed - Isabel Fiel M, Thung SN, Hytiroglou P, Emre S, Schiano TD. Liver failure and need for liver transplantation in patients with advanced hepatoportal sclerosis. Am J Surg Pathol. 2007;31(4):607-614.
doi pubmed - Krishnan P, Fiel MI, Rosenkrantz AB, Hajdu CH, Schiano TD, Oyfe I, Taouli B. Hepatoportal sclerosis: CT and MRI appearance with histopathologic correlation. AJR Am J Roentgenol. 2012;198(2):370-376.
doi pubmed - Levison DA, Kingham JG, Dawson AM, Stansfeld AG. Slow cirrhosis - or no cirrhosis? a lesion causing benign intrahepatic portal hypertension. J Pathol. 1982;137(3):253-272.
doi pubmed - Kingham JG, Levison DA, Stansfeld AG, Dawson AM. Non-cirrhotic intrahepatic portal hypertension: A long term follow-up study. Q J Med. 1981;50(199):259-268.
- Eapen CE, Nightingale P, Hubscher SG, Lane PJ, Plant T, Velissaris D, Elias E. Non-cirrhotic intrahepatic portal hypertension: associated gut diseases and prognostic factors. Dig Dis Sci. 2011;56(1):227-235.
doi pubmed - Ohbu M, Okudaira M, Watanabe K, Kaneko S, Takai T. Histopathological study of intrahepatic aberrant vessels in cases of noncirrhotic portal hypertension. Hepatology. 1994;20(2):302-308.
doi pubmed - Rajesh S, Mukund A, Sureka B, Bansal K, Ronot M, Arora A. Non-cirrhotic portal hypertension: an imaging review. Abdom Radiol (NY). 2018;43(8):1991-2010.
doi pubmed - Gioia S, Nardelli S, Pasquale C, Pentassuglio I, Nicoletti V, Aprile F, Merli M, et al. Natural history of patients with non cirrhotic portal hypertension: Comparison with patients with compensated cirrhosis. Dig Liver Dis. 2018;50(8):839-844.
doi pubmed - Nicoara-Farcau O, Rusu I, Stefanescu H, Tantau M, Badea RI, Procopet B. Diagnostic challenges in non-cirrhotic portal hypertension - porto sinusoidal vascular disease. World J Gastroenterol. 2020;26(22):3000-3011.
doi pubmed - Gioia S, Nardelli S, Ridola L, Riggio O. Causes and management of non-cirrhotic portal hypertension. Curr Gastroenterol Rep. 2020;22(12):56.
doi pubmed - Goel A, Ramakrishna B, Madhu K, Zachariah U, Ramachandran J, Keshava SN, Elias E, et al. Idiopathic noncirrhotic intrahepatic portal hypertension is an ongoing problem in India. Hepatology. 2011;54(6):2274; author reply 2274-2275.
doi pubmed - Sherlock S, Feldman CA, Moran B, Scheuer PJ. Partial nodular transformation of the liver with portal hypertension. Am J Med. 1966;40(2):195-203.
doi - Wanless IR, Lentz JS, Roberts EA. Partial nodular transformation of liver in an adult with persistent ductus venosus. Review with hypothesis on pathogenesis. Arch Pathol Lab Med. 1985;109(5):427-432.
- Steiner PE. Nodular regenerative hyperplasia of the liver. Am J Pathol. 1959;35:943-953.
- Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology. 1990;11(5):787-797.
doi pubmed - Radomski JS, Chojnacki KA, Moritz MJ, Rubin R, Armenti VT, Wilson GA, Herrine S, et al. Results of liver transplantation for nodular regenerative hyperplasia. Am Surg. 2000;66(11):1067-1070.
- Austin A, Campbell E, Lane P, Elias E. Nodular regenerative hyperplasia of the liver and coeliac disease: potential role of IgA anticardiolipin antibody. Gut. 2004;53(7):1032-1034.
doi pubmed - Malamut G, Ziol M, Suarez F, Beaugrand M, Viallard JF, Lascaux AS, Verkarre V, et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol. 2008;48(1):74-82.
doi pubmed - Leung VK, Loke TK, Luk IS, Ng WL, Chau TN, Law ST, Chan JC. Nodular regenerative hyperplasia of the liver associated with systemic lupus erythematosus: three cases. Hong Kong Med J. 2009;15(2):139-142.
- Jharap B, van Asseldonk DP, de Boer NK, Bedossa P, Diebold J, Jonker AM, Leteurtre E, et al. Diagnosing nodular regenerative hyperplasia of the liver is thwarted by low interobserver agreement. PLoS One. 2015;10(6):e0120299.
doi pubmed - Polish E, Christie J, Cohen A, Sullivan B, Jr. Idiopathic presinusoidal portal hypertension (Banti's syndrome). Ann Intern Med. 1962;56:624-627.
doi pubmed - Sciot R, Staessen D, Van Damme B, Van Steenbergen W, Fevery J, De Groote J, Desmet VJ. Incomplete septal cirrhosis: histopathological aspects. Histopathology. 1988;13(6):593-603.
doi pubmed - Valla DC. Primary Budd-Chiari syndrome. J Hepatol. 2009;50(1):195-203.
doi pubmed - Valla DC. Budd-Chiari syndrome/hepatic venous outflow tract obstruction. Hepatol Int. 2018;12(Suppl 1):168-180.
doi pubmed - Valla DC, Cazals-Hatem D. Vascular liver diseases on the clinical side: definitions and diagnosis, new concepts. Virchows Arch. 2018;473(1):3-13.
doi pubmed - Kakar S, Kamath PS, Burgart LJ. Sinusoidal dilatation and congestion in liver biopsy: is it always due to venous outflow impairment? Arch Pathol Lab Med. 2004;128(8):901-904.
doi pubmed - Lee H, Stark K, Fiel MI. Vascular pattern of injury. In: Liu X, Lai J, Jhala N, eds. Practical Interpretation of liver biopsy. Volume 1. Newcastle Upon Tyne: Cambridge Scholars Publishing, 2020:375-434.
- Valla DC, Cazals-Hatem D. Sinusoidal obstruction syndrome. Clin Res Hepatol Gastroenterol. 2016;40(4):378-385.
doi pubmed - DeLeve LD, Valla DC, Garcia-Tsao G, American Association for the Study Liver D. Vascular disorders of the liver. Hepatology. 2009;49(5):1729-1764.
doi pubmed - Saadoun D, Cazals-Hatem D, Denninger MH, Boudaoud L, Pham BN, Mallet V, Condat B, et al. Association of idiopathic hepatic sinusoidal dilatation with the immunological features of the antiphospholipid syndrome. Gut. 2004;53(10):1516-1519.
doi pubmed - Marzano C, Cazals-Hatem D, Rautou PE, Valla DC. The significance of nonobstructive sinusoidal dilatation of the liver: Impaired portal perfusion or inflammatory reaction syndrome. Hepatology. 2015;62(3):956-963.
doi pubmed - Bernard O, Franchi-Abella S, Branchereau S, Pariente D, Gauthier F, Jacquemin E. Congenital portosystemic shunts in children: recognition, evaluation, and management. Semin Liver Dis. 2012;32(4):273-287.
doi pubmed - Guido M, Sarcognato S, Sacchi D, Colloredo G. Pathology of idiopathic non-cirrhotic portal hypertension. Virchows Arch. 2018;473(1):23-31.
doi pubmed - Bissonnette J, Rautou PE, Valla DC. [Idiopathic non-cirrhotic portal hypertension: An update]. Presse Med. 2015;44(10):1009-1015.
doi pubmed - Wanless IR, Huang WY. Vascular disorders. In: Burt A, Portmann B, Ferrell L, eds. MacSween's pathology of the liver, 6th ed. Philadelphia: Churchill Livingstone Elsevier. 2012. 602-644.
- Batts KP, Ludwig J. Chronic hepatitis. An update on terminology and reporting. Am J Surg Pathol. 1995;19(12):1409-1417.
doi pubmed - Crawford JM. Liver cirrhosis. In: MacSween RNM, Burt AD, Portmann BC, et al, eds. Pathology of the Liver. 4th ed. Philadelphia: Harcourt, 2002:575-619.
- Hano H, Takasaki S. Three-dimensional observations on the alterations of lobular architecture in chronic hepatitis with special reference to its angioarchitecture for a better understanding of the formal pathogenesis of liver cirrhosis. Virchows Arch. 2003;443(5):655-663.
doi pubmed - Pulvirenti F, Pentassuglio I, Milito C, Valente M, De Santis A, Conti V, d'Amati G, et al. Idiopathic non cirrhotic portal hypertension and spleno-portal axis abnormalities in patients with severe primary antibody deficiencies. J Immunol Res. 2014;2014:672458.
doi pubmed - Agrawal M, Rahmani R, Nakkala K, Fiel MI, Schiano T. Hepatoportal sclerosis (obliterative portal venopathy) and nodular regenerative hyperplasia in a patient with myasthenia gravis: A case report and review of the published work. Hepatol Res. 2013;43(9):999-1003.
doi pubmed - Intagliata NM, Caldwell SH, Tripodi A. Diagnosis, Development, and Treatment of Portal Vein Thrombosis in Patients With and Without Cirrhosis. Gastroenterology. 2019;156(6):1582-1599 e1581.
doi pubmed - Scoazec JY, Marche C, Girard PM, Houtmann J, Durand-Schneider AM, Saimot AG, Benhamou JP, et al. Peliosis hepatis and sinusoidal dilation during infection by the human immunodeficiency virus (HIV). An ultrastructural study. Am J Pathol. 1988;131(1):38-47.
- Mallet VO, Varthaman A, Lasne D, Viard JP, Gouya H, Borgel D, Lacroix-Desmazes S, et al. Acquired protein S deficiency leads to obliterative portal venopathy and to compensatory nodular regenerative hyperplasia in HIV-infected patients. AIDS. 2009;23(12):1511-1518.
doi pubmed - Koot BG, Alders M, Verheij J, Beuers U, Cobben JM. A de novo mutation in KCNN3 associated with autosomal dominant idiopathic non-cirrhotic portal hypertension. J Hepatol. 2016;64(4):974-977.
doi pubmed - Vilarinho S, Sari S, Yilmaz G, Stiegler AL, Boggon TJ, Jain D, Akyol G, et al. Recurrent recessive mutation in deoxyguanosine kinase causes idiopathic noncirrhotic portal hypertension. Hepatology. 2016;63(6):1977-1986.
doi pubmed - Roulot D. Liver involvement in Turner syndrome. Liver Int. 2013;33(1):24-30.
doi pubmed - de Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta. 2009;1792(9):841-843.
doi pubmed - Vispo E, Cevik M, Rockstroh JK, Barreiro P, Nelson M, Scourfield A, Boesecke C, et al. Genetic determinants of idiopathic noncirrhotic portal hypertension in HIV-infected patients. Clin Infect Dis. 2013;56(8):1117-1122.
doi pubmed - Baffy G. Origins of portal hypertension in nonalcoholic fatty liver disease. Dig Dis Sci. 2018;63(3):563-576.
doi pubmed - Ryou M, Stylopoulos N, Baffy G. Nonalcoholic fatty liver disease and portal hypertension. Explor Med. 2020;1:149-169.
doi pubmed - Francque S, Wamutu S, Chatterjee S, Van Marck E, Herman A, Ramon A, Jung A, et al. Non-alcoholic steatohepatitis induces non-fibrosis-related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int. 2010;30(3):365-375.
doi pubmed - Francque S, Verrijken A, Mertens I, Hubens G, Van Marck E, Pelckmans P, Van Gaal L, et al. Noncirrhotic human nonalcoholic fatty liver disease induces portal hypertension in relation to the histological degree of steatosis. Eur J Gastroenterol Hepatol. 2010;22(12):1449-1457.
doi pubmed - Mendes FD, Suzuki A, Sanderson SO, Lindor KD, Angulo P. Prevalence and indicators of portal hypertension in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2012;10(9):1028-1033 e1022.
doi pubmed - Barbera A, Raurell I, Garcia-Lezana T, Torres-Arauz M, Bravo M, Hide D, Gil M, et al. Steatosis as main determinant of portal hypertension through a restriction of hepatic sinusoidal area in a dietary rat nash model. Liver Int. 2020;40(11):2732-2743.
doi pubmed - Yip WW, Burt AD. Alcoholic liver disease. Semin Diagn Pathol. 2006;23(3-4):149-160.
doi pubmed - Van der Graaff D, Kwanten WJ, Couturier FJ, Govaerts JS, Verlinden W, Brosius I, D'Hondt M, et al. Severe steatosis induces portal hypertension by systemic arterial hyporeactivity and hepatic vasoconstrictor hyperreactivity in rats. Lab Invest. 2018;98(10):1263-1275.
doi pubmed - Chawla Y, Dilawari JB. Anorectal varices - their frequency in cirrhotic and non-cirrhotic portal hypertension. Gut. 1991;32(3):309-311.
doi pubmed - Schouten JN, Nevens F, Hansen B, Laleman W, van den Born M, Komuta M, Roskams T, et al. Idiopathic noncirrhotic portal hypertension is associated with poor survival: results of a long-term cohort study. Aliment Pharmacol Ther. 2012;35(12):1424-1433.
doi pubmed - Rush N, Saxena R. Vascular disorders of the liver. In: Saxena R, ed. Practical hepatic pathology: A diagnostic approach. 2nd ed. Philadelphia, PA: Elsevier, 2018:467-484.
doi - Ma J, Gong X, Luo J, Gu J, Yan Z, Zhang W, Li C, et al. Impact of intrahepatic venovenous shunt on hepatic venous pressure gradient measurement. J Vasc Interv Radiol. 2020;31(12):2081-2088.
doi pubmed - Elkrief L, Lazareth M, Chevret S, Paradis V, Magaz M, Blaise L, Rubbia-Brandt L, et al. Liver stiffness by transient elastography to detect porto-sinusoidal vascular liver disease with portal hypertension. Hepatology. 2020.
doi - Futagawa S, Fukazawa M, Musha H, Isomatsu T, Koyama K, Ito T, Horisawa M, et al. Hepatic venography in noncirrhotic idiopathic portal hypertension. Comparison with cirrhosis of the liver. Radiology. 1981;141(2):303-309.
doi pubmed - Delladetsima I, Sakellariou S, Kokkori A, Tiniakos D. Atrophic hepatocytes express keratin 7 in ischemia-associated liver lesions. Histol Histopathol. 2016;31(10):1089-1094.
- Liang J, Shi C, Dupont WD, Salaria SN, Huh WJ, Correa H, Roland JT, et al. Key histopathologic features in idiopathic noncirrhotic portal hypertension: an interobserver agreement study and proposal for diagnostic criteria. Mod Pathol. 2021;34(3):592-602.
doi pubmed - Sarin SK, Wadhawan M, Gupta R, Shahi H. Evaluation of endoscopic variceal ligation (EVL) versus propanolol plus isosorbide mononitrate/nadolol (ISMN) in the prevention of variceal rebleeding: comparison of cirrhotic and noncirrhotic patients. Dig Dis Sci. 2005;50(8):1538-1547.
doi pubmed - Sarin SK, Gupta N, Jha SK, Agrawal A, Mishra SR, Sharma BC, Kumar A. Equal efficacy of endoscopic variceal ligation and propranolol in preventing variceal bleeding in patients with noncirrhotic portal hypertension. Gastroenterology. 2010;139(4):1238-1245.
doi pubmed - de Franchis R, Baveno VIF. Expanding consensus in portal hypertension: Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J Hepatol. 2015;63(3):743-752.
doi pubmed - Bissonnette J, Garcia-Pagan JC, Albillos A, Turon F, Ferreira C, Tellez L, Nault JC, et al. Role of the transjugular intrahepatic portosystemic shunt in the management of severe complications of portal hypertension in idiopathic noncirrhotic portal hypertension. Hepatology. 2016;64(1):224-231.
doi pubmed - European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Vascular diseases of the liver. J Hepatol. 2016;64(1):179-202.
doi pubmed - Woran K, Semmler G, Jachs M, Simbrunner B, Maria Bauer DJ, Binter T, Pomej K, et al. Clinical Course of Porto-Sinusoidal Vascular Disease Is Distinct From Idiopathic Noncirrhotic Portal Hypertension. Clin Gastroenterol Hepatol. 2020.
doi pubmed - Sood A, Cox GA, 2nd, McWilliams JP, Wang HL, Saab S. Patients with nodular regenerative hyperplasia should be considered for hepatocellular carcinoma screening. Hepatol Res. 2014;44(6):689-693.
doi pubmed - Nzeako UC, Goodman ZD, Ishak KG. Hepatocellular carcinoma and nodular regenerative hyperplasia: possible pathogenetic relationship. Am J Gastroenterol. 1996;91(5):879-884.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.