

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website http://www.gastrores.org |
Original Article
Volume 13, Number 1, February 2020, pages 32-39
Histologic Lesions of Porto-Sinusoidal Vascular Disease Following Phlebotomy in Hemochromatosis
Tony El Jabboura, Kelsey E. McHughb, Deepa T. Patilb, c, Chunlai Zuoa, Brandon H. Kood, Sungeun Kima, Hwajeong Leea, e
aAnatomic Pathology, Albany Medical College, Albany, NY, USA
bAnatomic Pathology, Cleveland Clinic, Cleveland, OH, USA
cPathology, Brigham and Women’s hospital, Boston, MA, USA
dAlbany Medical College, Albany, NY, USA
eCorresponding Author: Hwajeong Lee, Department of Pathology and Laboratory Medicine, Albany Medical Center, 47 New Scotland Ave., MC81, Albany, NY 12208, USA
Manuscript submitted October 19, 2019, accepted November 12, 2019
Short title: Histologic Lesions of PSVD Following Phlebotomy
doi: https://doi.org/10.14740/gr1236
| Abstract | ▴Top |
Background: Phlebotomy induces regression of liver fibrosis in genetic hemochromatosis. We assessed the histologic changes in pre-phlebotomy and post-phlebotomy liver biopsies from patients with HFE mutation as a model to study regression of fibrosis. We aimed to show that phlebotomy-induced histologic lesions overlap with porto-sinusoidal vascular disease (PSVD, also known as idiopathic non-cirrhotic portal hypertension), histologically.
Methods: A total of 51 biopsies (22 pre-phlebotomy and 29 post-phlebotomy) were reviewed, and three variables were studied: iron index indicative of the amount of accumulated iron (range 0 to 18), the combined score of vascular changes reflecting the presence of histological lesions that are described in PSVD (range 0 to 9) and the high-grade shunt vessel by calculating the proportion of portal tracts with shunt vessels, with a cutoff of 50%. Two-tailed Student’s t-test and Fisher’s exact test were performed to compare the means of two variables and frequencies of the histologic lesions in two groups, respectively. A P-value < 0.05 was considered statistically significant.
Results: The iron index was higher in the pre-phlebotomy compared to post-phlebotomy group (P = 0.01). Compared to the pre-phlebotomy group, the combined score was higher in the post-phlebotomy group when the cases of advanced fibrosis were excluded (P = 0.023) and remained higher when patients with risk factors for PSVD were further excluded (P = 0.034). The high-grade shunt vessel tended to be more common in the post-phlebotomy group when advanced fibrosis was excluded; however, the statistical significance was marginal (P = 0.056).
Conclusions: Phlebotomy reduces hepatic iron load and induces histologic lesions of PSVD in patients with HFE mutation. Our data support a postulation that some of the histologic lesions of PSVD represent vascular remodeling following a regression of fibrosis and may not be reflective of risk factors or etiopathogenesis of PSVD. Regressed fibrosis and PSVD may not be reliably distinguished in a limited sample, therefore warranting cautious interpretation in the right clinical context.
Keywords: Porto-sinusoidal vascular disease; Portal hypertension; Phlebotomy; Hemochromatosis; Fibrosis; Liver
| Introduction | ▴Top |
Progression of hepatic fibrosis is a result of continuous remodeling in response to chronic insult to the liver. In the past, cirrhosis was considered an irreversible end stage of hepatic injury [1]. However, recent studies have shown that advanced fibrosis and cirrhosis is reversible when the underlying liver condition causing the injury is treated. Examples include reversal of liver fibrosis in chronic hepatitis B virus (HBV) and hepatitis C virus (HCV)-induced hepatitis following treatment with interferon and/or nucleoside analogues and antivirals [2-6], and down-staging of fibrosis of autoimmune hepatitis following immunosuppressive treatment [7]. Similarly, serial liver biopsies from genetic hemochromatosis patients showed regression of fibrosis following phlebotomy treatment [8].
Idiopathic non-cirrhotic portal hypertension (INCPH) is a clinicopathologic entity in which liver samples from patients with clinical evidence of portal hypertension fail to show cirrhosis in the absence of other causes of portal hypertension [9-13]. Recently, the Vascular Liver Disease Interest Group (VALDIG) proposed a novel terminology “porto-sinusoidal vascular disease (PSVD)” in order to broaden the definition of INCPH and capture the earlier phase of the disease preceding portal hypertension [14]. Histologically, PSVD shows subtle vascular changes and associated histologic findings such as obliterative portal venopathy, nodular regenerative hyperplasia, incomplete septal fibrosis, portal tract abnormalities, architectural disturbance, non-zonal sinusoidal dilatation and mild perisinusoidal fibrosis, in the absence of cirrhosis [14]. Common risk factors include infectious etiology, hypercoagulability, rheumatologic and immunologic conditions, exposure to toxin/drug and genetic predisposition [9-14].
Compared to the description of subtle and variable histologic lesions of PSVD, histologic overlap between regressed fibrosis and PSVD in liver biopsy is less well documented in the literature. Dr. Wanless and other authors postulated that incomplete septal cirrhosis, a histologic lesion of PSVD [15, 16], represents a form of regressed fibrosis [17, 18].
Therefore, regression of fibrosis may pose a diagnostic challenge to clinicians and pathologists when a pertinent clinical history of chronic hepatitis, advanced fibrosis and treatment is unavailable, or when the histologic overlap between regressed fibrosis and PSVD is not recognized. Some of these biopsies may be interpreted as featuring histologic lesions of PSVD, which may lead to an unnecessary search for risk factors of PSVD, while underlying chronic hepatitis and possible regression of fibrosis are overlooked.
We recently encountered a liver biopsy from a patient with a past history of genetic hemochromatosis and cirrhosis. The biopsy showed neither parenchymal iron deposits nor fibrosis. The biopsy was remarkable for variable histologic lesions of PSVD, including an increased number of portal venous branches in portal tracts, obliterative portal venopathy, sinusoidal dilatation and nodular regeneration of the lobules. Further clinical history of regular phlebotomy was noted, and these histologic changes were attributed to regression of fibrosis following phlebotomy.
We hypothesize that review of pre-phlebotomy and post-phlebotomy liver samples of genetic hemochromatosis patients may provide direct histologic evidence of regressed fibrosis that is within the spectrum of PSVD. While the optimal study material would consist of sequential pre-phlebotomy and post-phlebotomy liver biopsies from a hemochromatosis patient cohort, such a cohort is unavailable at our institutions. Therefore, as an alternative, we studied liver biopsies from patients with known HFE gene mutation and compared pre-phlebotomy and post-phlebotomy biopsies. The goal of the study was to compare pre-phlebotomy and post-phlebotomy liver samples from HFE mutation cohort and evaluate histologic changes that can be seen in PSVD.
Unfortunately, there is no standardized method to quantify the degree or amount of individual histologic lesions pertaining to PSVD that correlates with clinical signs of portal hypertension [14, 19]. Also, it is unknown how many of these individual histologic features or any combination thereof, is required to make a histologic diagnosis of PSVD or predict portal hypertension. Therefore, as previously devised and utilized [10, 11], the individual histologic lesions of PSVD were evaluated for the absence (score 0) or presence (score 1) of the lesions, and the combined score (CS) of the individual lesions was assessed.
| Materials and Methods | ▴Top |
The study was approved by the Institutional Review Boards at the Albany Medical Center and Cleveland Clinic, respectively. This study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration. A random search for patients who underwent liver biopsy or resection while carrying a diagnosis of hemochromatosis was performed, using International Classification of Diseases, Ninth and Tenth Revision (ICD-9 and -10). For outpatients, the Current Procedural Terminology (CPT) code was used.
Mutational status of C282Y and H63D, demographics and phlebotomy history were obtained from the electronic medical records. Cases of secondary/acquired siderosis without genetic abnormality were excluded. All available liver biopsies and resections from the patients were retrieved.
Archived hematoxylin and eosin (H&E), trichrome, Prussian blue and periodic acid-Schiff-diastase (PAS/D) stained slides prepared from the formalin-fixed, paraffin-embedded tissue blocks at the time of the diagnoses were reviewed by two (TEJ and HL) pathologists, blinded to the phlebotomy history.
The number of portal tracts was counted and the degree of iron accumulation (see iron index below) and METAVIR fibrosis stage were evaluated. The individual histologic lesions of PSVD were evaluated for the absence or presence of the lesions, as previously devised [10, 11] (Table 1 [10, 20]) (Fig. 1). The degree of shunt vessel (SV) was further quantified by enumerating the number of portal tracts with SVs in a given biopsy (see high-grade SV below). Due to overlapping histologic features between fatty liver disease (FLD) and PSVD [10], histologic features of FLD were evaluated when present.
 Click to view | Table 1. Histological Features of Liver Biopsies and Definition [10, 20] |
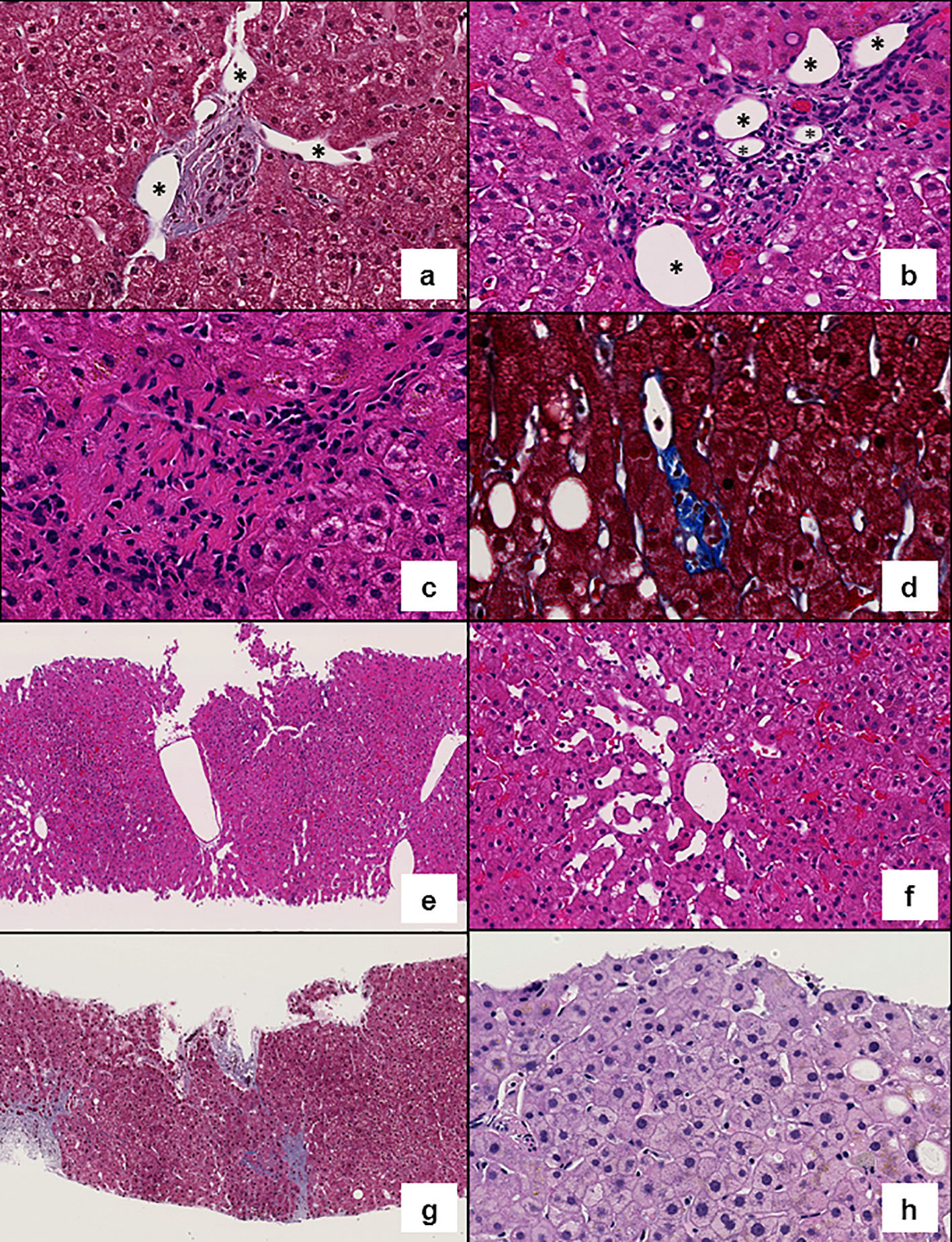 Click for large image | Figure 1. Representative images of histologic features evaluated. (a) Shunt vessels (*) herniating into the liver parenchyma (Masson’s trichrome, × 200). (b) Increased number of portal vessels: multiple portal vein branches (*) are present in a portal tract (hematoxylin and eosin (H&E), × 200). (c) Obliterative portal venopathy (H&E, × 300). (d) Rudimentary portal tract: hypoplastic portal tract in which the lumen of the bile duct is smaller than the diameter of the surrounding hepatocytes (Masson’s trichrome, × 300). (e) Central vein clustering (H&E, × 100). (f) Sinusoidal dilatation (H&E, × 200). (g) Incomplete fibrous septa (Masson’s trichrome, × 100). (h) Anisocytosis (H&E, × 200). |
The above data were used to generate three quantifiable variables for the pre-phlebotomy and post-phlebotomy group: the CS of vascular changes, the iron index and the frequency of high-grade SV. Two-tailed Student’s t-test was performed when comparing the means of two groups. Fisher’s exact test was performed when comparing the frequencies of specific histologic features in two groups. A P value of less than 0.05 was considered statistically significant.
CS of vascular changes
A score of 0 or 1 was assigned for the absence or presence of P3, P4, P5, P6, L1, L2, L5, L6 and L8. A CS ranging from 0 to 9 was generated by summing the score of the histologic features for each biopsy, as previously used [10] (Table 1). The mean of those scores was calculated for both the pre-phlebotomy and post-phlebotomy groups.
Iron index
This index was devised to assign a quantitative value for iron accumulation. An iron granularity (G) score of 1, 2, 3, 4, 5 or 6 was assigned when the iron granules were seen at × 40, × 20, × 10, × 4, × 2, or grossly on the glass slide by the naked eye, respectively. An iron zonation (Z) score of 1, 2 and 3 was assigned when the iron granules were seen in hepatic zone 1, zones 1 and 2, and zones 1 to 3, respectively. In the absence of iron, both G and Z scores are 0. The iron index was defined as the G score multiplied by the Z score (G × Z). The mean of iron indices was calculated for both pre-phlebotomy and post-phlebotomy groups.
High-grade SV
A biopsy was considered to have high-grade SV when ≥ 50% of its portal tracts exhibited SVs. The percentage of pre-phlebotomy and post-phlebotomy biopsies with high-grade SV was calculated.
| Results | ▴Top |
Demographics and hemochromatosis gene mutation
A total of 41 patients who met the study criteria were included in the analysis. There were 28 males and 13 females, with a mean age of 54 years, ranging from 17 to 83 years. Twenty-nine patients were homozygotes for C282Y mutation, nine were homozygotes for H63D mutation, two were C282Y/H63D heterozygotes and one was a C282Y carrier. Twelve patients had one or more potential risk factors of PSVD, including immunologic dysregulation, rheumatologic disease, chronic venous outflow obstruction, malignancy, infection and exposure to drug. No patient had portal hypertension at the time of liver biopsy or resection.
Pre-phlebotomy and post-phlebotomy groups
Fifteen of 29 C282Y homozygotes, three of nine H63D homozygotes, both two C282Y/H63D heterozygotes and the C282Y carrier had undergone phlebotomy at the time of first liver biopsy. A set of pre-phlebotomy and post-phlebotomy biopsy was available in five patients. In two additional patients, two or more serial post-phlebotomy biopsies were available for review. In total, 51 biopsies and resections were available for review, of which 22 were pre-phlebotomy and 29 were post-phlebotomy specimens.
In the pre-phlebotomy group, the biopsy was performed at the time of genetic hemochromatosis diagnosis in 11 cases, within 1 - 2 months of diagnosis in seven cases, and at 10 years in one case. One patient had a liver biopsy 4.5 years prior to the diagnosis of hereditary hemochromatosis. The time of biopsy in relation to diagnosis was unknown in two patients.
In the post-phlebotomy group, the biopsy was done within 1 year of diagnosis in four cases, 3 years in two cases, 5 - 10 years in nine cases, 10 - 20 years in seven cases and > 20 years in three cases. The time of biopsy in relation to diagnosis was unknown in four cases. In this group, phlebotomy was performed at variable frequencies, ranging from weekly to every 6 months. Some patients underwent regular phlebotomies, whereas the rest did not.
Eight out of 22 (36%) cases in the pre-phlebotomy group and eight out of 29 cases (28%) in the post-phlebotomy group demonstrated advanced fibrosis (METAVIR F3 and F4); frequencies of advanced fibrosis did not statistically differ between these two groups. Co-existing FLD was noted in 14 out of 22 (64%) pre-phlebotomy and 14 out of 29 (48%) post-phlebotomy specimens; frequencies of FLD were not statistically different between these two groups. Three out of 22 (14%) pre-phlebotomy and 10 of 29 (34%) post-phlebotomy cases showed incomplete fibrous septa (L5) on trichrome stain. The difference in the frequencies of incomplete fibrous septa was statistically insignificant. Incomplete fibrous septa without centrizonal perisinusoidal fibrosis were seen only in two post-phlebotomy biopsies.
CS of vascular changes
The CS of vascular changes was not statistically different between pre-phlebotomy (n = 22) and post-phlebotomy (n = 29) groups (3.4 (pre-phlebotomy) vs. 3.9 (post-phlebotomy); P = 0.160). However, exclusion of the cases with advanced fibrosis (METAVIR F3, F4) in both groups led to a remarkable increase of the CS in the post-phlebotomy group compared to the pre-phlebotomy group (3.6 (pre-phlebotomy, n = 14) versus 4.8 (post-phlebotomy, n = 21); P = 0.023) with a statistical significance. When patients with potential risk factors for PSVD were further excluded, the CS in both groups remained unchanged, and statistical significance was maintained (3.6 (pre-phlebotomy, n = 9) versus 4.8 (post-phlebotomy, n = 18); P = 0.034).
Iron index
This index was higher in the pre-phlebotomy group when compared to the post-phlebotomy group (8.3 versus 4.3) with a statistical significance (P = 0.01).
High-grade SV
After cases with advanced fibrosis (METAVIR F3, F4) were excluded, only one of 14 (7%) pre-phlebotomy group cases showed high-grade SV, whereas it was present in eight of 21 (38%) post-phlebotomy group cases. However, statistical significance was marginal with a P value of 0.056 (Table 2).
Click to view | Table 2. Summary of Results |
Serial liver biopsies
Five sets of pre-phlebotomy and post-phlebotomy biopsies from five patients, and two sets of serial post-phlebotomy biopsies from two patients were available. Among these, two patients that developed hepatocellular carcinoma in the second biopsies, one patient that had a progression of severe FLD and one that had persistent FLD were excluded from evaluation. Therefore, three sets of pre-phlebotomy and post-phlebotomy biopsies from three patients without confounding factors were finally evaluated. The duration of phlebotomy was 4 years and 9 months, 1 year, and 3 years and 2 months, respectively.
In these three patients, compared to pre-phlebotomy biopsies, post-phlebotomy biopsies showed reduction of METAVIR fibrosis stage (F2 to F0, F3 to F2, and F2 to F0), and increased CS of vascular changes (3 to 7, 5 to 7, and 3 to 8). High-grade SV was present only in one post-phlebotomy biopsy in the third patient.
| Discussion | ▴Top |
PSVD is a clinicopathologic entity of the liver comprised of subtle vascular lesions and associated histologic changes without histologic evidence of cirrhosis [14]. These vascular and associated histologic changes are relatively well documented in PSVD with portal hypertension (previously known as INCPH). However, their significance in the absence of clinical portal hypertension - PSVD without portal hypertension - remains unclear. This may encompass heterogeneous entities with differing etiopathogenesis [10, 11, 14, 21]. The histologic lesions of PSVD were described in patients with and without risk factors for PSVD, in pediatric patients, in FLD, in autopsy liver, in liver biopsies obtained intraoperatively during cholecystectomies and gastric bypasses, and in trauma patients without portal hypertension [10, 11, 21-25]. Regardless of their clinical annotation, histologic lesions of PSVD appear to represent a transient phase or an end result of heterogeneous perfusion, in association with obliterative portal venopathy, regeneration and remodeling of the liver [10, 11, 22].
It has also been recognized that regression of liver fibrosis induces vascular changes that can be seen in PSVD. Wanless et al carried out a meticulous morphometric evaluation of regression of fibrosis in liver samples from serial biopsies and 52 liver explants, and showed that regression of fibrosis from micro- and macronodular cirrhosis to incomplete septal cirrhosis results in variable histologic changes such as thin fibrous septa, rudimentary portal tracts, regenerative nodules and aberrant arrangement of hepatic veins [17]. Schinoni et al described regenerative nodules, hypoplastic portal tracts, periportal fibrosis, altered lobular architecture, phlebosclerosis and SVs (described as dilated thin-walled vessels in the interface between the portal tract and parenchyma) in their study of incomplete septal cirrhosis [18]. These histologic lesions are well documented in PSVD [9, 12-14]. Likewise, Sciot et al documented similarities in histologic features between incomplete septal cirrhosis and PSVD suggesting a common etiopathogenesis, while the authors considered incomplete septal cirrhosis as a subtype of cirrhosis [15].
We chose an HFE mutation cohort to study histologic changes related to regression of fibrosis due to phlebotomy’s known role in the regression of fibrosis in genetic hemochromatosis [8], and because hepatic iron accumulation is a quantifiable histologic variable that can be used to confirm the effect of phlebotomy. As anticipated, the iron index was higher in the pre-phlebotomy group than in the post-phlebotomy group. This confirms that the effect of phlebotomy represented by a lower iron index is evident in the post-phlebotomy group, while the frequencies of advanced fibrosis and FLD are not statistically different from those in the pre-phlebotomy group, and validates our study design.
We have shown that histologic lesions that can be seen in PSVD are more pronounced in the post-phlebotomy group than in the pre-phlebotomy group, especially when confounding factors such as advanced fibrosis are excluded. Similarly, although the P value was marginal, the frequency of high-grade SV in the post-phlebotomy group tended to be higher than that of the pre-phlebotomy group. These results support the notion that phlebotomy leads to vascular and histologic changes that resemble PSVD in patients with hemochromatosis gene mutation.
Falize et al looked at the METAVIR fibrosis stage of pre-phlebotomy and post-phlebotomy biopsies from genetic hemochromatosis patients. The authors showed that among 36 patients with pre-phlebotomy stage of F3 or F4, 21 patients demonstrated reduction in fibrosis following phlebotomy, of which 14 patients showed minimal fibrosis (F0 to F1) in post-phlebotomy biopsies. Interestingly, one patient in their study showed cirrhosis (METAVIR F4) in the initial biopsy and no fibrosis (F0) in the post-phlebotomy biopsy [8]. While the authors did not evaluate histologic features of PSVD in their study, and despite having only three patients with serial biopsies without significant confounding factors in our study, the post-phlebotomy biopsies in our study showed down-staging of the METAVIR fibrosis score and higher combined vascular scores than those in the pre-phlebotomy biopsies. These observations further support the notion that regression and resorption of fibrosis may be associated with PSVD-like histologic changes in liver biopsy.
Given the overlapping histologic features between regressed fibrosis and PSVD, from a histologic standpoint, distinguishing between these two would be extremely challenging unless pertinent histories and clinical contexts are provided and the histologic overlap is recognized. Even so, recognition of regressed fibrosis or incomplete septal cirrhosis appears to be difficult especially in biopsy samples [15]. In Wanless et al’s study regarding regression of fibrosis, the samples were mainly from explant specimens except from one patient with serial core needle biopsies [17]. Likewise, Ibarrola et al successfully identified one case of incomplete septal cirrhosis out of nine PSVD cases. However, the specimens from this study were from autopsies and liver resections [16]. Incomplete septal cirrhosis associated with Wegener’s granulomatosis was also diagnosed on autopsy [26]. As the presence of incomplete fibrous septa with no perisinusoidal fibrosis appeared specific for incomplete septal cirrhosis in Ibarrola’s study, we looked at the frequency of incomplete fibrous septa (L5) in our cases. There was a trend of increased frequency of incomplete fibrous septa in the post-phlebotomy group. However, statistical significance was not reached, and most of these cases had co-existing perisinusoidal fibrosis due to superimposed FLD.
We agree with previous postulations that regression of fibrosis appears to represent a transient phase of a dynamic process consisting of repeated injury, scarring and resorption of fibrosis. This continuous remodeling appears to be associated with, or conducive to heterogeneous perfusion of the liver and obliterative portal venopathy. Any type of chronic hepatitis would undergo this continuous remodeling process. In his original definition of incomplete septal cirrhosis, Dr. Popper noticed that this is frequently associated with alcoholic and viral hepatitis [27]. Subsequently, incomplete septal cirrhosis was reported in HBV hepatitis, HCV hepatitis, alcoholic hepatitis, alpha-1 antitrypsin deficiency, Wegener’s granulomatosis, following high-dose methylprednisolone and as idiopathic [17, 26, 28]. Similarly, Schinoni et al performed a histomorphologic study of eight incomplete septal cirrhosis cases and reported that five of eight patients had known chronic hepatitis due to autoimmune disease, alcohol and HCV. The samples showed varying stages of progression and regression of inflammation and fibrosis, suggesting that incomplete septal cirrhosis is an intermediate stage of the remodeling process [18]. Our observation in this study that phlebotomy induces PSVD-like histologic lesions, likely by resorption of fibrosis and remodeling, further supports this postulation.
The limitations of our study include a relatively low case number after exclusion of METAVIR F3 and F4 cases, and frequent co-existence with FLD that confounded histologic evaluation. However, despite the low case number, statistical significance was maintained before and after excluding cases with risk factors for PSVD in CS calculation. Also, the degree of iron overload and baseline liver injury was uneven. For example, a relatively young patient (17-year-old) and six patients with H63D homozygosity were included in the pre-phlebotomy group. However, the iron index was lower in the post-phlebotomy group compared to that in the pre-phlebotomy group, and there were three patients with H63D homozygosity in the post-phlebotomy group. Moreover, a minor subset of patients with H63D mutation may manifest clinically significant iron overload [29]. Therefore, we believe that the study design was adequate in addressing the study goal. Lastly, whether hepatocellular anisocytosis can be used as a surrogate for nodular regeneration or not is to be validated.
There are no standardized histopathologic diagnostic criteria to render a diagnosis of PSVD in a liver biopsy. Some authors devised grading schemes or arbitrary histologic criteria for their studies and applied them to cases of PSVD with portal hypertension; however, the grading schemes and proposed diagnostic criteria did not correlate with clinical outcome including signs of portal hypertension, hemodynamic status and coagulability [19, 30]. The diagnosis of PSVD is further compounded by high interobserver variability [31, 32]. The scoring scheme we devised and used for the previous studies and current study is our attempt to address the subtle and somewhat subjective histologic changes of PSVD in a simple and systemic manner [10, 11].
Conclusion
Liver biopsy interpretation plays a crucial role in rendering a diagnosis of PSVD. When subtle histologic features of PSVD are encountered in liver biopsies without portal hypertension in patients with any form of chronic hepatitis or liver injury, ongoing remodeling process may account for the histologic lesions. When these features are seen in the absence of known chronic hepatitis, liver injury or signs of portal hypertension, some of them would fulfill the recently proposed diagnostic criteria for “PSVD without portal hypertension”, with risk factors for PSVD in a subset [11, 14]. When there is a clinical history of portal hypertension in biopsies featuring histologic lesions of PSVD without cirrhosis, a diagnosis of “PSVD with portal hypertension” would be justified, however, with a caveat that some of these biopsies may represent regressed cirrhosis and may be unrelated to risk factors of PSVD.
Acknowledgments
None to declare.
Financial Disclosure
The authors received no financial support for the research, authorship and/or publication of this article.
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
The authors made substantial contributions to the conception and design of the work (HL), the acquisition, analysis, or interpretation of data for the work (TEJ, KEM, DTP, CZ, BHK, SK, HL), drafting the work (TEJ, HL), and revised it critically for important intellectual content (KEM, DTP, CZ, BHK, SK). The authors approve final version that is to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
| References | ▴Top |
- Hytiroglou P, Theise ND. Regression of human cirrhosis: an update, 18 years after the pioneering article by Wanless et al. Virchows Arch. 2018;473(1):15-22.
doi pubmed - Dienstag JL, Goldin RD, Heathcote EJ, Hann HW, Woessner M, Stephenson SL, Gardner S, et al. Histological outcome during long-term lamivudine therapy. Gastroenterology. 2003;124(1):105-117.
doi pubmed - Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT, Kitis G, Rizzetto M, Marcellin P, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology. 2006;131(6):1743-1751.
doi pubmed - Chang TT, Liaw YF, Wu SS, Schiff E, Han KH, Lai CL, Safadi R, et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology. 2010;52(3):886-893.
doi pubmed - Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, Washington MK, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;381(9865):468-475.
doi - Poynard T, McHutchison J, Manns M, Trepo C, Lindsay K, Goodman Z, Ling MH, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122(5):1303-1313.
doi pubmed - Czaja AJ, Carpenter HA. Decreased fibrosis during corticosteroid therapy of autoimmune hepatitis. J Hepatol. 2004;40(4):646-652.
doi pubmed - Falize L, Guillygomarc'h A, Perrin M, Laine F, Guyader D, Brissot P, Turlin B, et al. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology. 2006;44(2):472-477.
doi pubmed - Schouten JN, Garcia-Pagan JC, Valla DC, Janssen HL. Idiopathic noncirrhotic portal hypertension. Hepatology. 2011;54(3):1071-1081.
doi pubmed - Zuo C, Chumbalkar V, Ells PF, Bonville DJ, Lee H. Prevalence of histological features of idiopathic noncirrhotic portal hypertension in general population: a retrospective study of incidental liver biopsies. Hepatol Int. 2017;11(5):452-460.
doi pubmed - Lee H, Ainechi S, Singh M, Ells PF, Sheehan CE, Lin J. Histological Spectrum of Idiopathic Noncirrhotic Portal Hypertension in Liver Biopsies From Dialysis Patients. Int J Surg Pathol. 2015;23(6):439-446.
doi pubmed - Lee H, Rehman AU, Fiel MI. Idiopathic Noncirrhotic Portal Hypertension: An Appraisal. J Pathol Transl Med. 2016;50(1):17-25.
doi pubmed - Aggarwal S, Fiel MI, Schiano TD. Obliterative portal venopathy: a clinical and histopathological review. Dig Dis Sci. 2013;58(10):2767-2776.
doi pubmed - De Gottardi A, Rautou PE, Schouten J, Rubbia-Brandt L, Leebeek F, Trebicka J, Murad SD, et al. Porto-sinusoidal vascular disease: proposal and description of a novel entity. Lancet Gastroenterol Hepatol. 2019;4(5):399-411.
doi - Sciot R, Staessen D, Van Damme B, Van Steenbergen W, Fevery J, De Groote J, Desmet VJ. Incomplete septal cirrhosis: histopathological aspects. Histopathology. 1988;13(6):593-603.
doi pubmed - Ibarrola C, Colina F. Clinicopathological features of nine cases of non-cirrhotic portal hypertension: current definitions and criteria are inadequate. Histopathology. 2003;42(3):251-264.
doi pubmed - Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med. 2000;124(11):1599-1607.
- Schinoni MI, Andrade Z, de Freitas LA, Oliveira R, Parana R. Incomplete septal cirrhosis: an enigmatic disease. Liver Int. 2004;24(5):452-456.
doi pubmed - Verheij J, Schouten JN, Komuta M, Nevens F, Hansen BE, Janssen HL, Roskams T. Histological features in western patients with idiopathic non-cirrhotic portal hypertension. Histopathology. 2013;62(7):1083-1091.
doi pubmed - Pewzner-Jung Y, Brenner O, Braun S, Laviad EL, Ben-Dor S, Feldmesser E, Horn-Saban S, et al. A critical role for ceramide synthase 2 in liver homeostasis: II. insights into molecular changes leading to hepatopathy. J Biol Chem. 2010;285(14):10911-10923.
doi pubmed - Cazals-Hatem D, Hillaire S, Rudler M, Plessier A, Paradis V, Condat B, Francoz C, et al. Obliterative portal venopathy: portal hypertension is not always present at diagnosis. J Hepatol. 2011;54(3):455-461.
doi pubmed - Wanless IR, Bernier V, Seger M. Intrahepatic portal vein sclerosis in patients without a history of liver disease. An autopsy study. Am J Pathol. 1982;106(1):63-70.
- Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology. 1990;11(5):787-797.
doi pubmed - Cantez MS, Gerenli N, Ertekin V, Gulluoglu M, Durmaz O. Hepatoportal sclerosis in childhood: descriptive analysis of 12 patients. J Korean Med Sci. 2013;28(10):1507-1511.
doi pubmed - Nakanuma Y, Hoso M, Sasaki M, Terada T, Katayanagi K, Nonomura A, Kurumaya H, et al. Histopathology of the liver in non-cirrhotic portal hypertension of unknown aetiology. Histopathology. 1996;28(3):195-204.
doi pubmed - Zen Y, Sunagozaka H, Tsuneyama K, Masutomi K, Terasaki S, Kaneko S, Kobayashi K, et al. Incomplete septal cirrhosis associated with Wegener's granulomatosis. Liver. 2002;22(5):388-393.
doi pubmed - Popper H. What are the major types of hepatic cirrhosis? In: Ingelfinger F, Relman A, Finland M, ed. Controversy in Internal Medicine. Philadelphia: Saunders, 1996: p. 223-243.
- Grilli E, Galati V, Petrosillo N, Del Nonno F, Baiocchini A. Incomplete septal cirrhosis after high-dose methylprednisolone therapy and regression of liver injury. Liver Int. 2015;35(2):674-676.
doi pubmed - Heeney MM, Andrews NC. Iron hemeostasis and inherited iron overload disorders: an overview. Hematol Oncol Clin North Am. 2004;18(6):1-20.
doi pubmed - Hillaire S, Bonte E, Denninger MH, Casadevall N, Cadranel JF, Lebrec D, Valla D, et al. Idiopathic non-cirrhotic intrahepatic portal hypertension in the West: a re-evaluation in 28 patients. Gut. 2002;51(2):275-280.
doi pubmed - Jharap B, van Asseldonk DP, de Boer NK, Bedossa P, Diebold J, Jonker AM, Leteurtre E, et al. Diagnosing nodular regenerative hyperplasia of the liver is thwarted by low interobserver agreement. PLoS One. 2015;10(6):e0120299.
doi pubmed - Zuo C, Lin J, Affolter K, Choi W-T, Lagana SM, Liu X, KE Choi, et al. Histologic overlap in liver biopsies from patients with and without non-cirrhotic portal hypertension (NCPH): Interobserver agreement study. Lab Investig. 2019;99(Suppl 1):61-62.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.