

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website https://www.gastrores.org |
Case Report
Volume 16, Number 4, August 2023, pages 244-248

Coma With Hyperammonemia in a Patient With Kwashiorkor
Thibault Vieillea, d, Francois Feilletb, c, Arnaud Wiedemannb, c, Hadrien Winiszewskia, Gael Pitona
aService de Reanimation Medicale, CHRU de Besancon, Boulevard Fleming, 25030 Besancon, France
bCentre de Reference des Maladies Metaboliques, Service de Pediatrie, CHRU de Nancy, 54000 Nancy, France
cInserm UMR_S 1256, Nutrition Genetique et Exposition aux Risques Environnementaux (NGERE), Faculte de Medecine de Nancy, Universite de Lorraine, 54000 Nancy, France
dCorresponding Author: Thibault Vieille, Service de Reanimation Medicale, CHRU de Besancon, Boulevard Fleming, 25030 Besancon, France
Manuscript submitted May 7, 2023, accepted July 3, 2023, published online August 26, 2023
Short title: Coma With Hyperammonemia and Kwashiorkor
doi: https://doi.org/10.14740/gr1634
| Abstract | ▴Top |
We describe a case of coma-related hyperammonemia in a woman presenting with severe edematous malnutrition (Kwashiorkor-like), without underlying hepatic disease. Our main hypothesis is that the patient developed a functional urea cycle disorder, due to the inability to synthesize N-acetylglutamate which is the activator of the first enzymes (carbamoyl phosphate synthetase) of urea cycle, in a context of severe deficiency of essential amino acids and of acetyl-CoA. Severe hyperammonemia is a medical emergency exposing to the risk of cerebral edema. Urgent treatment should interrupt protein intake, stimulate protein anabolism, and remove ammonia from the blood using renal replacement therapy and ammonia scavengers. Hyperammonemia should be searched in case of unexplained coma, even among patients without hepatic disorder, in particular among young patients. Hyperammonemia should also be searched among patients with severe protein-calorie malnutrition.
Keywords: Hyperammonemia; Coma; Urea cycle disorder; Malnutrition; Kwashiorkor
| Introduction | ▴Top |
We describe a case of coma-related hyperammonemia in a woman presenting with severe edematous malnutrition (Kwashiorkor-like), without underlying hepatic disease.
| Case Report | ▴Top |
A 35-year-old woman with personal history of depression, severe alcoholism complicated with polyneuropathy, heroin addiction, living in a context of social isolation, was admitted to the intensive care unit (ICU) for severe acute respiratory failure requiring mechanical ventilation.
One month before, she consulted her family physician for asthenia, general weakness, and inability to walk. She was hospitalized thereafter for 1 month in a general hospital. A diagnosis of severe malnutrition was performed. Interrogatory revealed the loss of 20 kg bodyweight in the last weeks. Physical examination highlighted development of diffuse edema of lower limbs. The patient presented with scaly and crusty cutaneous plaques on the back of the legs and popliteal fossae. Examination of buccal mucosa identified atrophic glossitis. The patient was evaluated by a dermatologist considering that cutaneous and mucosal lesions were related to severe malnutrition. Laboratory test revealed low albumin and prealbumin plasma concentration of 20 g/L (normal: 35 - 50 g/L) and 0.1 g/L (normal: 0.2 - 0.4 g/L), respectively. Enteral nutrition using a nasogastric tube was started. An accidental removal of the nasogastric tube was responsible for aspiration pneumonia and acute respiratory failure. The patient required orotracheal intubation and was then transferred to the ICU of our hospital.
At ICU admission, physical examination revealed many erythematous skin lesions with crusts on the back of the thighs and scratching skin lesions on the elbows, and diffuse edema of lower limbs. The patient presented with severe frontal and occipital alopecia. The body mass index was 23 kg/m2 but was largely overestimated due to diffuse edema. Biological variables at ICU admission are described in Table 1.
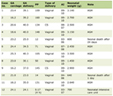 Click to view | Table 1. Description of Plasma Concentration of Biological Variables at ICU Admission |
Clinical course of respiratory failure was favorable, and enteral nutrition was started at ICU admission. However, awakening was delayed with presence of coma, anisocoria with reactive mydriasis, spastic hypertonia, abnormal extension after nociceptive stimulation, and manifestation of pyramidal syndrome with Babinski sign. Brain computed tomography (CT) scan was normal. Electroencephalogram revealed non-reactive and reduced cerebral activity, without sign of epilepsy or metabolic impairment. Abdominal CT revealed diffuse jejunitis, colitis and moderate ascites.
Finally, ammonia plasma concentration was found very increased at 185 µmol/L (normal: 18 - 72 µmol/L) (Fig. 1). In order to investigate this hyperammonemia, analysis of abdominal CT scan did not find pattern of chronic liver disease excepted for hepatic steatosis. Analyses of plasma amino acids by chromatography showed severe deficit of essential amino acids (tryptophane (< 5 µmol/L; normal: 20 - 60 µmol/L), valine, phenylalanine, methionine, leucine, isoleucine) as well as alanine, arginine, and citrulline (6 µmol/L; normal: 20 - 40 µmol/L) (Fig. 2). On the contrary, plasma glutamine concentration was increased (1,088 µmol/L; normal 400 - 800 µmol/L) showing functional deficiency of the urea cycle. Urinary orotic acid level was within the normal range, eliminating ornithine transcarbamylase (OTC) deficiency. Free carnitine and total plasma carnitine were above the normal range.
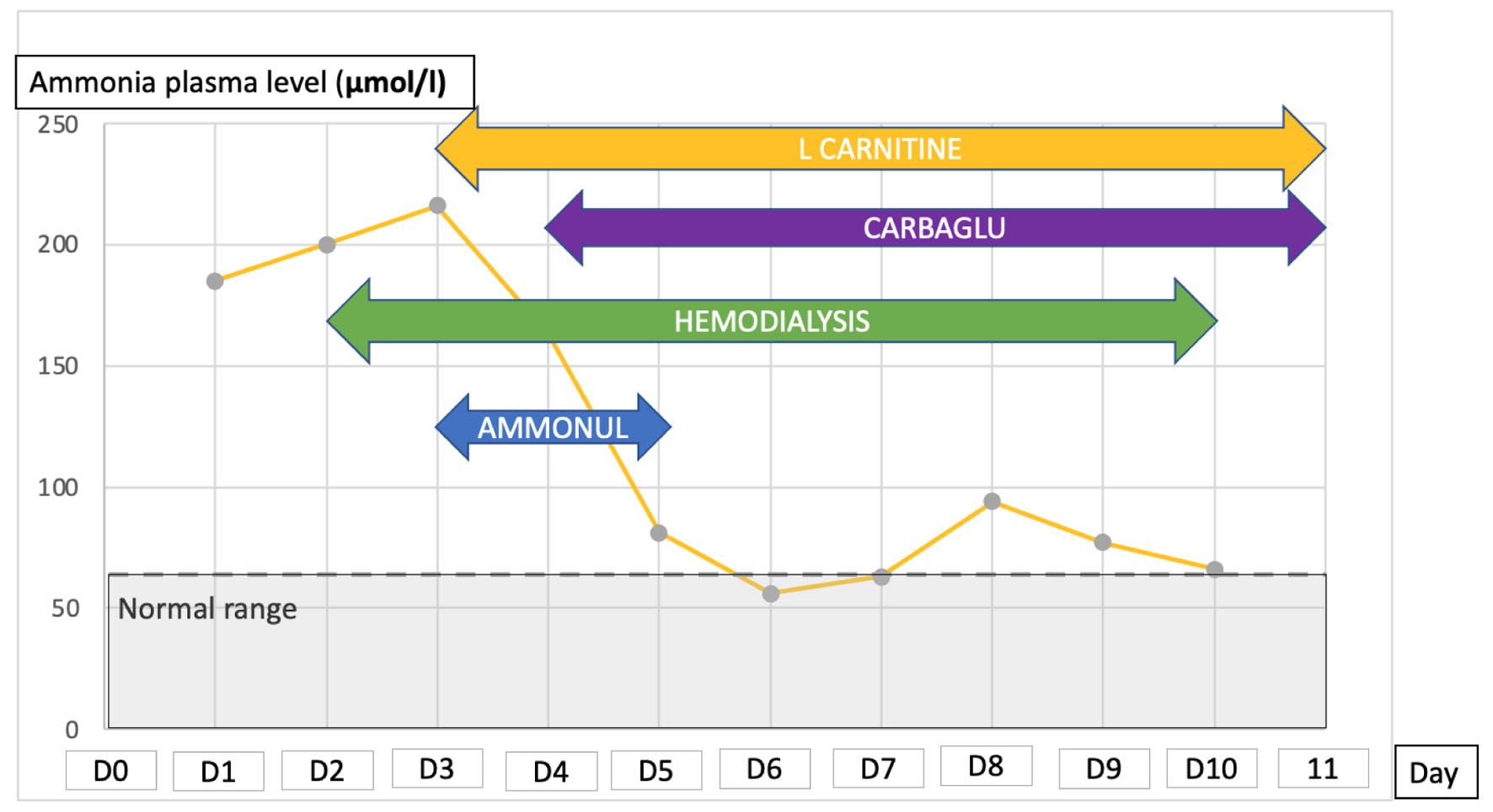 Click for large image | Figure 1. Evolution of plasma ammonia level during ICU stay and treatment for controlling the hyperammonemia. ICU: intensive care unit. |
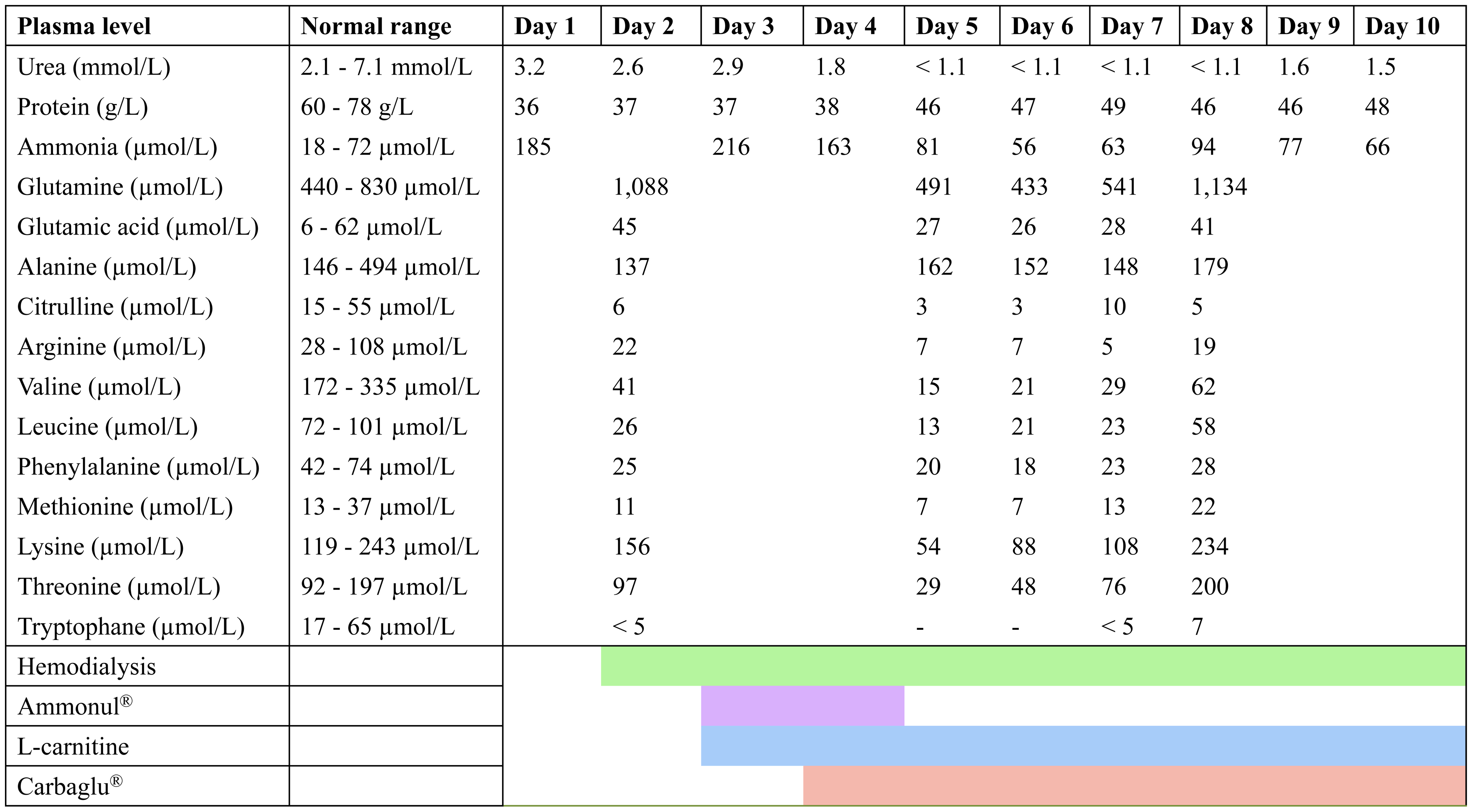 Click for large image | Figure 2. Evolution of Plasma Amino Acids Concentration During ICU Stay. |
Enteral protein intake was interrupted and continuous veno-venous hemodiafiltration (CVVHD) was started. Intravenous administration of sodium benzoate and sodium phenylacetate (AMMONUL©) was started and continued for 2 days. Thereafter, L-carnitine and carglumic acid (CARBAGLU©) were started and given for 1 week. CCVHD was continued for 1 week until control of ammonia. Clinical course of our patient was favorable. Ammonia plasma level reduced to normal quickly after starting management. Oral food with protein intake was then restarted. The patient was discharged from the ICU 12 days after the diagnosis of hyperammonemia with a normal neurological exam.
| Discussion | ▴Top |
This case illustrates a rare association of severe malnutrition with coma-associated hyperammonemia. We proposed the hypothesis that such coma-associated hyperammonemia is related to a functional urea cycle disorder, consequence of the Kwashiorkor.
The patient presented with clinical evidence of Kwashiorkor: history of severe protein energy malnutrition in a context of social isolation with severely limited food intake and persistent drug addiction, with significant body weight loss, generalized edema, ascites, skin lesions, alopecia, diffuse enteropathy at abdominal CT, severe hypoalbuminemia, essential amino acids deficiency, mainly tryptophan which was undetectable in plasma.
Orotracheal intubation was initially performed for acute respiratory failure, whereas no coma was described before. However, we identified a delayed awakening with persistent coma, whereas general anesthesia had been stopped for several days. The coma was associated with pupillary abnormalities and hypertonia, whereas the cerebral CT was normal. Among the causes of coma among severely malnourished patients, the diagnoses of refeeding syndrome, vitamin B1 deficiency, and hypoglycemia should be considered. In addition, hyperammonemia should be considered among patients presenting with coma without obvious cause, even in absence of known hepatic disease.
Ammonia (NH3+) is a product of amino acids catabolism. Urea cycle consists of several enzymatic reactions, predominantly in the liver, allowing to convert ammonia into urea, then excreted in the urine (Fig. 3). Urea cycle disorders result in the accumulation of ammonia, leading to hyperammonemia. The most common cause of hyperammonemia is severe liver disease. Sakusic et al described a series of 3,908 ICU patients with hyperammonemia. Among them, only 5% were not related to hepatic failure [1]. Other mechanisms may lead to hyperammonemia without underlying liver disease: acquired or inherited urea cycle disorders, and drugs toxicity (valproic acid or carbamazepine for example) [2]. Zao et al identified that hyperammonemia without liver disease was an independent factor of 30-day mortality in the ICU [2]. However, in the study by Sakusic et al, plasma level of ammonia was not associated with prognosis [1].
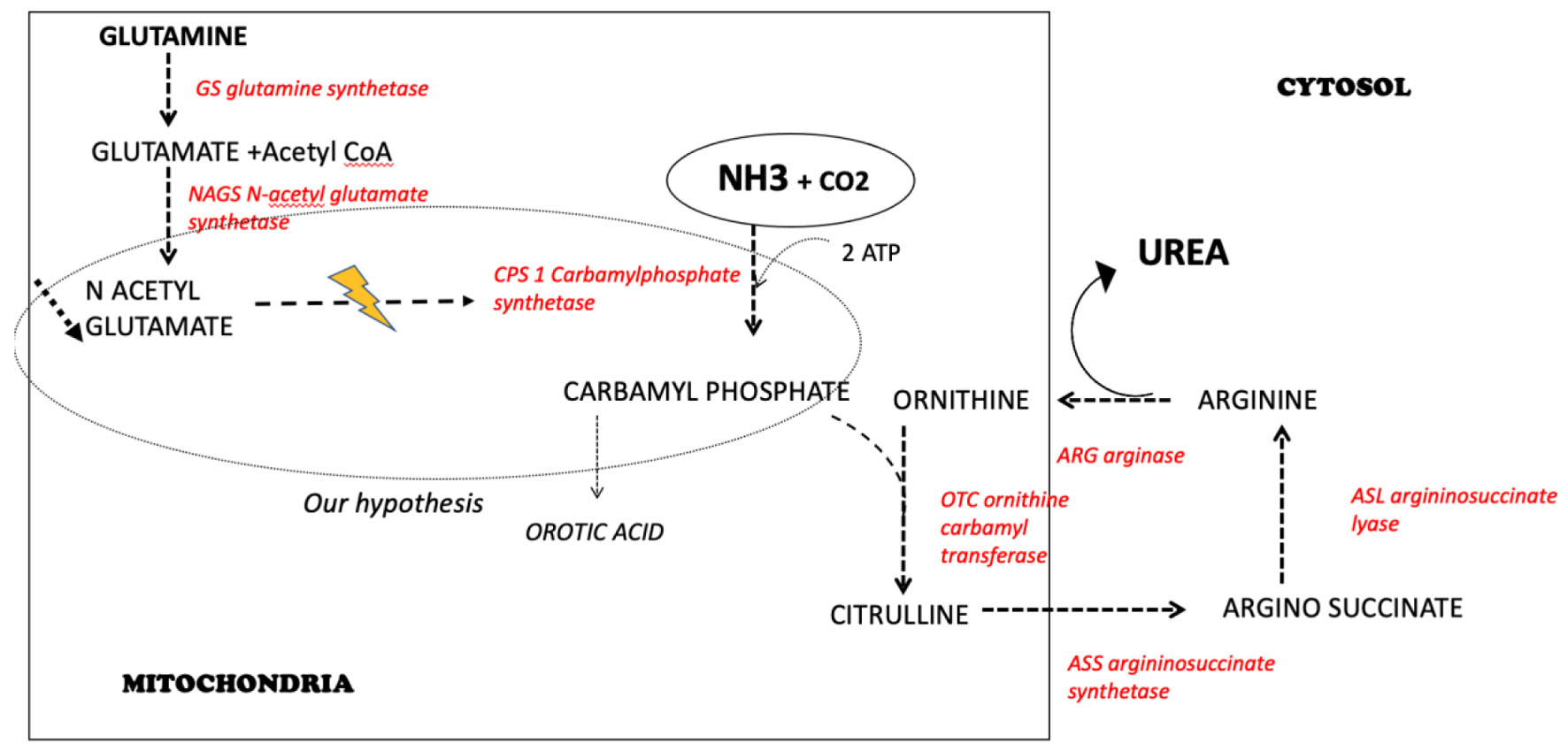 Click for large image | Figure 3. The urea cycle: our hypothesis to explain hyperammonemia related to a urea cycle disorder and a deficiency in N-acetylglutamate. |
Clinical manifestations of hyperammonemia are not specific: anorexia, vomiting, psychiatric symptom, altered level of consciousness from lethargy to coma, seizure, and hypothermia [3]. Life-threatening hyperammonemia is a medical emergency. The most important risk is the development of cerebral edema. Ammonia raises astrocyte glutamine synthesis responsible for glutamine accumulation leading to astrocyte swelling and cerebral hypertension [4]. Jacoby et al described a series of 78 patients presenting with hyperammonemia in the ICU (median plasma level 201 µmol/L). Hyperammonemia-related cerebral edema occurred in 8/78 patients, and 7/8 died [5]. The risk of brain edema could be particularly increased when plasma ammonia level is greater than 200 µmol/L [6]. A regular monitoring by transcranial Doppler looking for signs of intracranial hypertension and neuroprotective measures should be performed.
For the treatment of severe hyperammonemia, first of all, synthesis of ammonia should be reduced. For this purpose, all protein intakes should be withdrawn. In addition, the protein anabolism should be stimulated by supplying intravenous glucose and lipids. Secondly, ammonia should be removed from the body. Renal replacement therapy by CVVHD may allow clearance of ammonia. In addition, we should use pharmacological ammonia scavenging agents. Indeed sodium benzoate and sodium phenylacetate conjugate glycine and glutamine to form respectively hippuric acid and phenylacetylglutamine, both excreted in urine [7]. Finally, carglumic acid is a structural analog of N-acetylglutamate and activates carbamoyl-phosphate synthetase (CPS) to boost urea cycle, promoting ammonia detoxification.
How could we explain such hyperammonemia in this severely undernourished patient? Hyperammonemic encephalopathy has been described once in malnourished children [8].
In addition, Abo-Hussein et al showed that children with Kwashiorkor presented with hyperammonemia, and increased ammonia concentration in cerebrospinal fluid [9]. He proposed in 1984 the hypothesis that mental changes observed during Kwashiorkor could be related to hyperammonemia. Recently, a case of Kwashiorkor related to Roux-En-Y gastric bypass was described in an adult, and was associated with hyperammonemia at 136 mmol/L [10]. Our measurements documented a high plasma level of glutamine, and a reduced plasma essential amino acid level (Fig. 2). Glutamic acid level, plasma and urine orotic acid level were within the normal range, therefore excluding an OTC deficiency. Our main hypothesis is that this urea cycle disorder was related to a deficit in N-acetylglutamate which is the activator of CPS. This hypothesis is fitting with the metabolic profile associating high glutamine with normal orotic acid concentrations. Since a primary deficiency of CPS was very unlikely, the only possibility was a deficiency in N-acetylglutamate, and therefore we added carglumic acid in the treatment of our patient. A severe tryptophan deficiency induces a limitation in protein synthesis and is probably the cause of the severe protein deficiency. As it is shown in children Kwashiorkor, we observed a fatty liver due to a decreased metabolism of fatty acids and consecutively a deficiency in acetyl-CoA production.
N-acetylglutamate needs glutamate (not deficient in our patient) and acetyl-CoA to be synthetized by the enzyme N-acetylglutamate synthetase (NAGS). The metabolic pattern described above, and the good evolution of our patient are in favor of this pathophysiologic explanation. Our observation highlights the importance of using carglumic acid in addition to the current recommendations for hyperammonemia management in patients, mainly when they present with a severe malnutrition status. Severe protein deficiency, in particular the undetectable tryptophan plasma concentration could be responsible for functional urea cycle disorder, with the inability of urea cycle enzymes to be synthesized. In addition, since ammonia detoxification requires ATP, it has been described that a severe hypophosphatemia in a context of refeeding syndrome could be responsible for subsequent urea cycle disorder [11].
In conclusion, we describe a case of coma-related hyperammonemia in a woman presenting with Kwashiorkor syndrome. Our main hypothesis is that the patient developed a functional urea cycle disorder, due to N-acetylglutamate deficiency inducing an absence of stimulation of the CPS, the enzyme responsible for the first step of the urea cycle in a context of severe deficit of essential amino acids, mainly tryptophan. Severe hyperammonemia is a medical emergency exposing to the risk of cerebral edema. Urgent treatment should interrupt protein intake, stimulate protein anabolism, and remove ammonia from the blood using renal replacement therapy and ammonia scavengers. Carglumic acid should be systematically used in this context to optimize the urea cycle functioning.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
TV and GP drafted the manuscript. FF, AW, and HW have reviewed the manuscript and consistently improved its content. All authors read and approved the final manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Sakusic A, Sabov M, McCambridge AJ, Rabinstein AA, Singh TD, Mukesh K, Kashani KB, et al. Features of adult hyperammonemia not due to liver failure in the ICU. Crit Care Med. 2018;46(9):e897-e903.
doi pubmed pmc - Zhao L, Walline JH, Gao Y, Lu X, Yu S, Ge Z, Zhu H, et al. Prognostic role of ammonia in critical care patients without known hepatic disease. Front Med (Lausanne). 2020;7:589825.
doi pubmed pmc - Walker V. Severe hyperammonaemia in adults not explained by liver disease. Ann Clin Biochem. 2012;49(Pt 3):214-228.
doi pubmed - Takahashi H, Koehler RC, Brusilow SW, Traystman RJ. Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol. 1991;261(3 Pt 2):H825-829.
doi pubmed - Jacoby KJ, Singh P, Prekker ME, Leatherman JW. Characteristics and outcomes of critically ill patients with severe hyperammonemia. J Crit Care. 2020;56:177-181.
doi pubmed - Blei AT. Brain edema in acute liver failure. Crit Care Clin. 2008;24(1):99-114.
doi pubmed - Haberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32.
doi pubmed pmc - Morsy MR, Madina H, Sharaf SA, Soliman AT, Elzalabany MM, Ramadan MA. Hyperammonemia in Marasmic children. J Trop Pediatr. 1994;40(2):97-99.
doi pubmed - Abo-Hussein SA, Hussein ZM, Farag SI, Shebl SS, el-Melegy S, Akhnoukh S. A profile of ammonia-urea values in blood and cerebrospinal fluid in children with protein energy malnutrition. J Trop Med Hyg. 1984;87(6):237-240.
pubmed - Kim GN, Ho S, Saulino D, Liu X. Severe protein-calorie malnutrition-associated hepatic steatosis in a woman who had Roux-en-Y gastric bypass for morbid obesity thirteen years ago. Gastroenterology Res. 2021;14(2):129-137.
doi pubmed pmc - Becker S, Dam G, Hvas CL. Refeeding encephalopathy in a patient with severe hypophosphataemia and hyperammonaemia. Eur J Clin Nutr. 2015;69(2):279-281.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.