

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website http://www.gastrores.org |
Original Article
Volume 12, Number 1, February 2019, pages 8-15
The Influence of Hepatitis C Viral Loads on Natural Killer Cell Function
Mark Collistera, b, Cindy Ellisonb, c, Qian Lia, b, Gerald Y. Minukb, d, Julia D. Rempela, b, Sam K. Kunga
aDepartment of Immunology, University of Manitoba, Winnipeg, MB, Canada
bSection of Hepatology, University of Manitoba, Winnipeg, MB, Canada
cDepartment of Internal Medicine and Department of Pathology, University of Manitoba, Winnipeg, MB, Canada
dCorresponding Author: Gerald Y. Minuk, University of Manitoba, John Buhler Research Centre, 715 McDermot Ave., Winnipeg, MB R3E 3P4, Canada
Manuscript submitted August 30, 2018, accepted October 12, 2018
Short title: NK Cell Exhaustion in HCV Infection
doi: https://doi.org/10.14740/gr1081w
| Abstract | ▴Top |
Background: Hepatitis C virus (HCV) infection has a high rate of chronicity, attributable to its capacity to alter host immunity, including natural killer (NK) cell function. In this study, the interaction between NK cell activity and HCV viral load was investigated.
Methods: Peripheral blood NK cells were examined for cytotoxicity and interferon (IFN)-γ expression in HCV infected low (LVL, < 800,000 IU/mL, n = 10) and high (HVL, > 800,000 IU/mL, n = 13) viral load patient cohorts.
Results: Spontaneous NK cell cytotoxicity was more robust in the LVL cohort resulting in a negative correlation with viral loads (spontaneous, r = -0.437, P = 0.037; IFN-α activated, r = -0.372, P = 0.081). Although the percent of IFN-γ+ NK cells did not associate with viral load, within the LVL cohort there was a marked increase in IFN-γ+ NK cells upon IFN-α activation relative to medium alone (P < 0.01). To examine the inability of NK cells derived from HVL patients to be further activated, the expression of the exhaustion marker programmed cell death protein (PD)-1 was evaluated. PD-1 expression upon NK cell activation correlated with viral load (r = 0.649, P = 0.009). In addition, HCV proteins upregulated PD-1 expression in vitro (P < 0.05), suggesting that HCV can directly promote NK cell exhaustion. Cells from HVL patients were also more likely to produce IFN-γ in response to HCV core protein. The finding that NK cell PD-1 and IFN-γ expression are linked (r = 0.542, P < 0.05) suggests that increased IFN-γ levels may induce PD-1 as a negative feedback mechanism.
Conclusions: High HCV loads appear to promote NK exhaustion in chronic HCV infection.
Keywords: NK cells; Hepatitis C; Cytotoxicity; IFN-gamma; PD-1
| Introduction | ▴Top |
Hepatitis C virus (HCV) infection has a high rate of chronicity, with a global prevalence of approximately 2-3% [1]. Chronic HCV infection is a significant risk factor for progression to liver fibrosis and cancer [2]. Previous data indicate that, chronicity is maintained through viral-induced inhibition of protective host immune responses [3-5].
Natural killer (NK) cells are considered a primary target of HCV immune inhibition [6-8]. NK cells are innate immune cells that are critical in controlling viral replication [9, 10]. They act through cytotoxic killing of infected cells and the secretion of antiviral cytokines, most prominently interferon (IFN)-γ [11-13]. In vitro experiments indicate that HCV impairment of NK activity can occur at various levels [14]. For example, HCV envelope 2 protein can directly impair NK cell cytotoxic granule release and IL-2 induced IFN-γ synthesis through binding of cognate CD81 receptors [15, 16]. In addition, HCV virus can indirectly restrict NK cell activation by inhibiting dendritic cell secretion of IFN-α, a strong activator of NK cell cytotoxicity and IFN-γ production [17].
These in vitro findings are supported by clinical data. Patients with chronic HCV infection have lower numbers and percentages of NK cells in the peripheral blood compared to healthy individuals [15, 16, 18-20]. Whether this represents impaired NK cell proliferation or an increased NK cell migration into the liver is currently unknown [5]. Clinical studies further indicate that chronic HCV infection can affect NK effector functions. For example, NK cells from HCV infected patients have reduced cytotoxicity and IFN-γ production compared to cells from healthy controls [19, 21-23]. Moreover, Golden-Mason et al demonstrated that NK cell expression of programmed cell death protein (PD)-1 from HCV infected individuals was significantly higher in comparison to healthy control populations [24]. PD-1 expression has been linked to NK cell quiescence [25-27], but was originally described as an exhaustion marker on T cells upon cellular inertia in cancer and chronic viral infections including HCV infection [28-32]. Whether these interactions between NK cells and HCV are influenced by viral load has yet to be determined.
In the present study, we examined the effects of viral load on NK cell function and PD-1 expression in chronic HCV infected patients. We observed diminished NK cell activity with increasing viral load and which appeared to be a function of enhanced NK cell PD-1 expression.
| Patients and Methods | ▴Top |
Patient recruitment and viral loads
This study was approved by the University of Manitoba Research Ethics Board. Participants provided written informed consent. Treatment naive chronic HCV infected patients were recruited through the Viral Hepatitis Investigation Unit, Health Sciences Centre, Winnipeg, MB, Canada. Participants were HIV and hepatitis B core antibody negative. None were receiving immunosuppressive medications. Viral loads were measured by quantitative PCR at the Cadham Provincial Laboratory, MB, Canada by the COBAS® AmpliPrep/COBAS® TaqMan® HCV Quantitative Test, v2.0 assay (Roche Diagnostics Canada, Laval, QC).
NK cell cytotoxicity
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood with ficoll (Histopaque®, Sigma, St. Louis, MO) as previously described [33]. PBMC viability was determined using trypan blue exclusion. Cells consistently exhibited > 98% viability [33]. NK cell cytotoxicity was evaluated in standard 4 h chromium (Cr)51 release assays [34]. Fresh PBMCs were cultured overnight with, or without, recombinant human IFN-α2b (1,000 IU/mL, PBL InterferonSource, Piscataway, NJ). The following day, PBMCs were washed, added to a 96 well v-bottom plate (Corning) and serially diluted to achieve effector: target (E:T) ratios of 25:1, 12.5:1 and 6.25:1. K562 target cells, labeled with 50 µCi Na2CrO4 (PerkinElmer, Waltham, MA), were added at 1.0 × 104 cells/well. K562 cells are prototypic NK-specific target cells. Control wells to measure minimal, or maximal, release were established by, respectively, adding medium or 5% Triton X-100 (Sigma) directly to labeled target cells. After 4 h of incubation, plates were spun and counts per minute (cpm) were measured on a Wallac Wizard 1470 Gamma Counter (PerkinElmer). All analyses were done in triplicate. NK cell lysis was defined as the amount of Cr51 released into the medium upon target cell lysis. Percent specific lysis was calculated as ((cpm test - cpm spontaneous release)/(cpm max release - cpm spontaneous release)) × 100. NK cell lysis at the E:T ratio of 12.5:1 was used for analysis as this value consistently represented the linear portion of the lysis curve.
Culture stimulations with K562 cells
Intracellular cytokine staining (ICCS) was performed as previously described [35]. Briefly, frozen PBMC were thawed, washed and aliquoted (2.5 × 105 cells) into 5 mL round bottom tubes (BD Biosciences). Cells were incubated with brefeldin A in the absence, and presence, of K562 cells for 6 h. Fixable Viability Dye eFluor® 506 was added for 20 min on ice. Cells were then washed and fluorochrome-conjugated antibodies against CD56, CD3, IFN-γ and PD-1 were added for 30 min on ice. Appropriate isotype controls were included. PBMC were washed and fixed with 2% paraformaldehyde (Canemco Inc., Lakefield, Quebec). The following day data were acquired on a FACS Canto II (BD Biosciences, Mississauga, ON). Acquisition was performed using FACSDiva software and analysis was performed using FlowJo 7.6 (Tree Star Inc., Ashland, OR). For gating strategy see in Supplementary Figure 1 (www.gastrores.org).
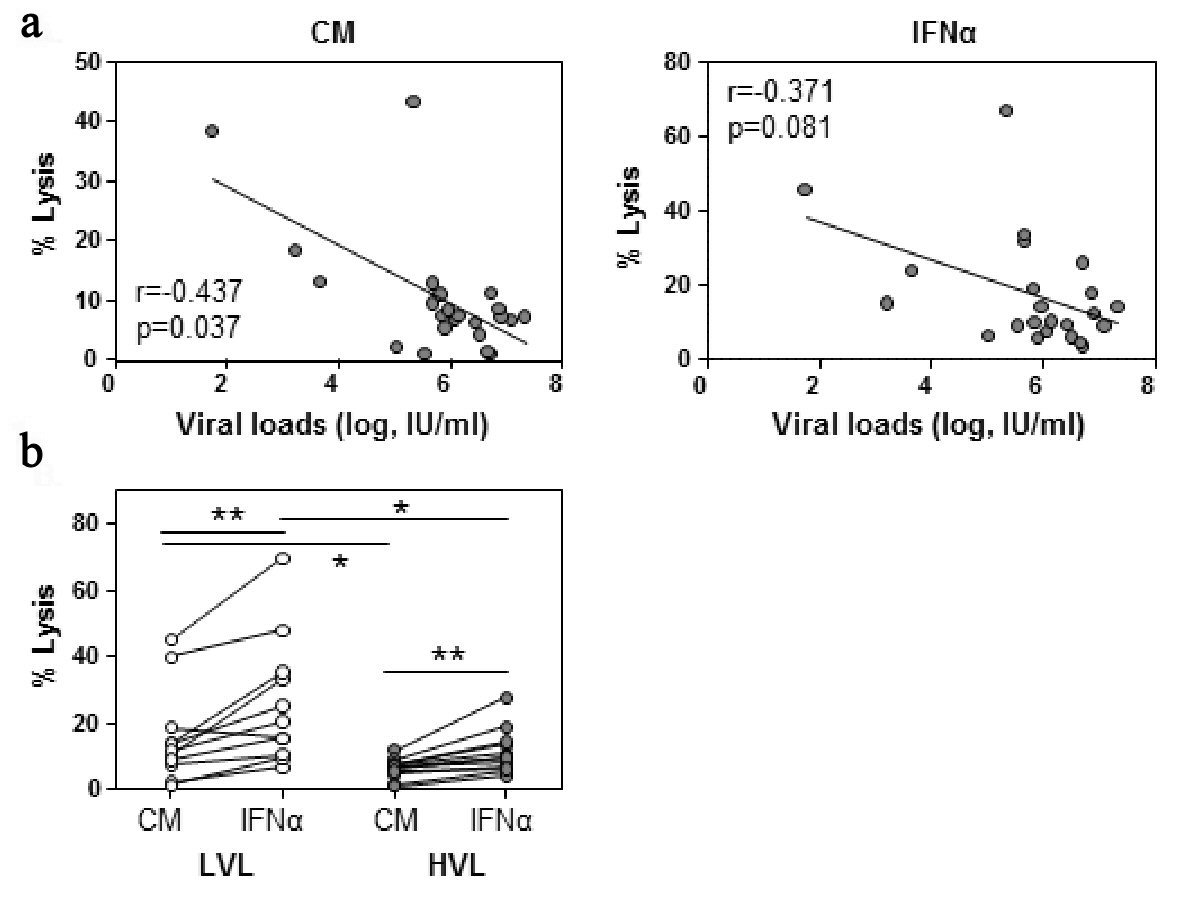 Click for large image | Figure 1. NK cell cytotoxicity decreases with viral load. PBMC were incubated overnight in culture medium (CM) and IFN-α. NK cell cytotoxicity was measured by Cr-51 release assay. (a) Baseline viral loads were plotted against NK cell cytotoxicity at an E:T ratio of 12.5:1 ratio following CM or IFN-α incubation. Spearman rank correlation, r, is shown. (b) Baseline NK cell cytotoxicity was evaluated between low viral load (LVL) and high viral load (HVL) patients. Lines indicate one patient’s values for each condition. Within cohort significant differences between CM and IFN-α were determined by paired t-test. Between cohorts significant differences were determined by Mann-Whitney U test. Significant differences are indicated as *P < 0.05, **P < 0.01. |
Culture stimulations with HCV proteins
PBMC were cultured in 10% (v/v) heat-inactivated fetal calf serum (Invitrogen Life Technologies, Grand Island, NY), RPMI 1640 (Hyclone, Logan, UT) medium. Cells were incubated with β-galactosidase (β-gal), β-gal linked HCV core (1 µg/mL), or 1 µg/mL HCV protein cocktail containing equivalent concentrations of core, nonstructural protein 3 (NS3), and NS4 or recombinant IL-2. Recombinant HCV core, NS3 and NS4 proteins corresponding to amino acids 2-192, 1450-1643, 1658-1863 respectively of the HCV polyprotein were received from Virogen (Watertown, MA). Cells were stained by for CD56, CD3, IFN-γ or PD-1 expression following 6 h culture (for gating strategy see supplementary Figures 2, 3 (www.gastrores.org)). Parallel cultures were incubated for 72 h, at which time supernatants were harvested and analyzed for IFN-γ levels by enzyme linked immunosorbent assay (ELISA) (BD Biosciences).
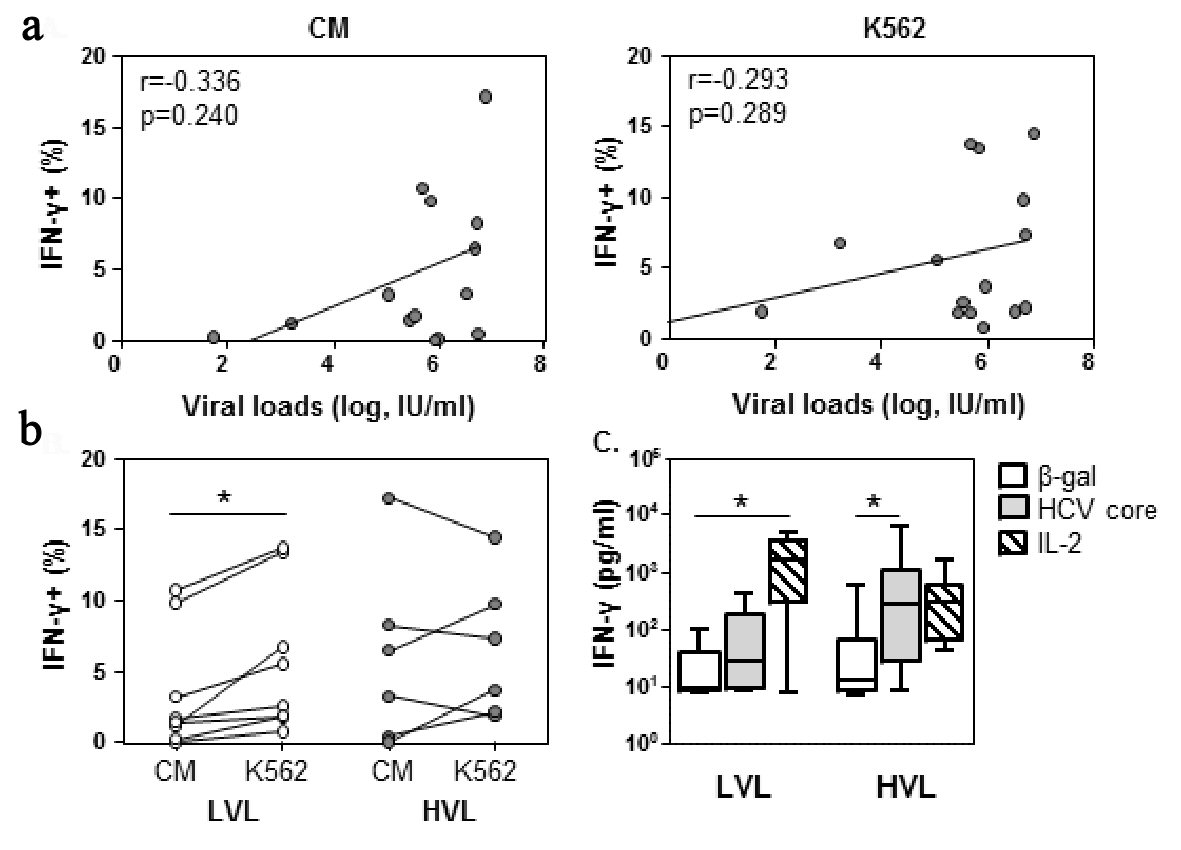 Click for large image | Figure 2. NK cells at low viral loads can be further stimulated towards IFN-γ expression by target cells. PBMC were incubated with CM or K562 target cells for 6 h in the presence of brefeldin A, and then stained for IFN-γ expression. (a) IFN-γ expression was assessed against viral load. Spearman rank correlation, r, is shown. P < 0.05 is considered significant. (b) The percent of NK cells producing IFN-γ was evaluated for LVL and HVL cohorts. Lines indicate an individuals values for each condition. Within cohort significant differences between CM and K562 were determined by paired t-test. Between cohorts significant differences were determined by Mann-Whitney U-test. (c) PBMCs were cultured with β-gal, core or IL-2 as indicated. Supernatants were collected at 72 h and analyzed for IFN-γ levels in LVL (n = 6) and HVL (n = 11). Statistical evaluation for changes in IFN-γ levels within a cohort was determined by ANOVA, repeated measures. Significant differences are indicated as *P < 0.05. |
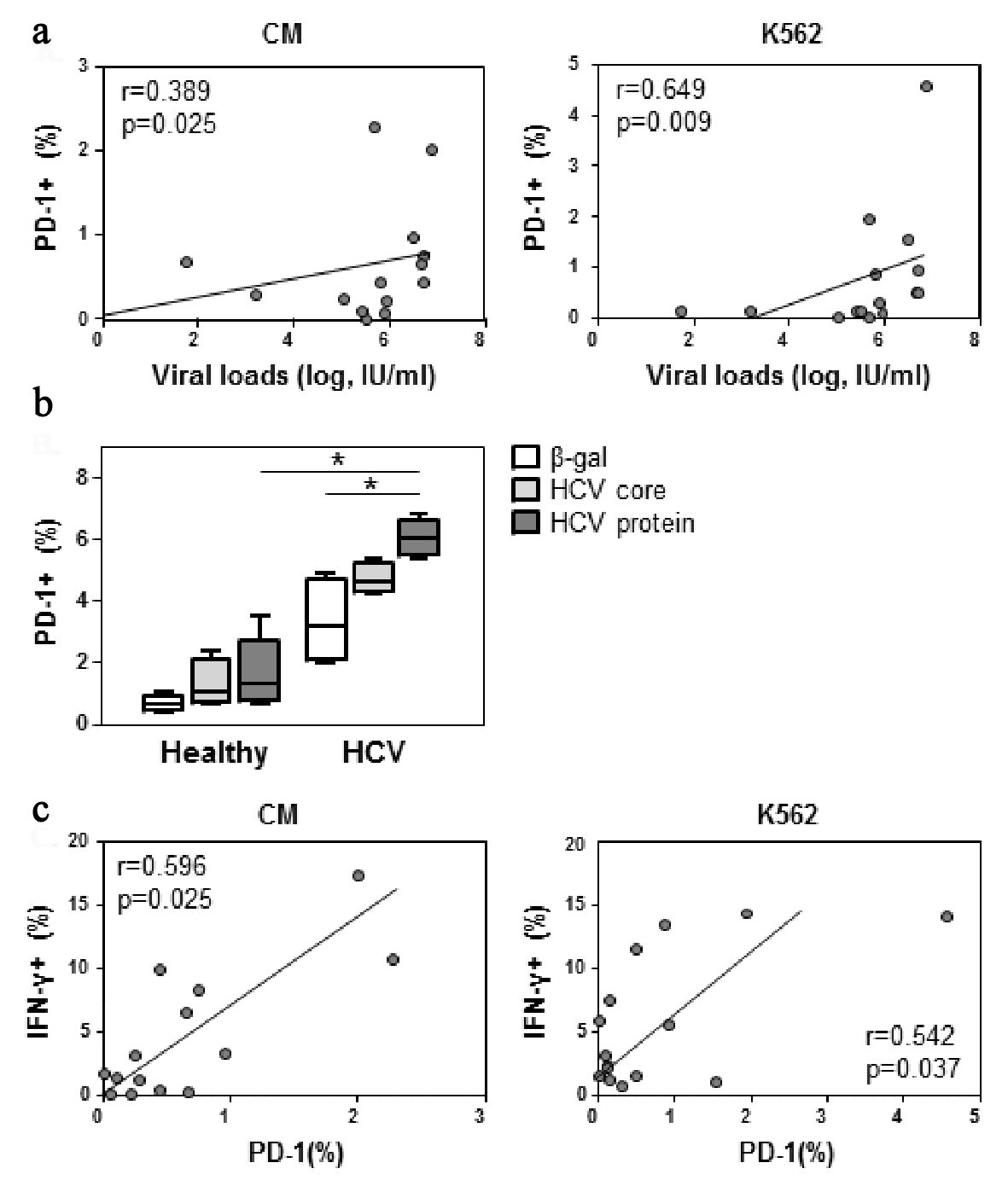 Click for large image | Figure 3. PD-1 expression correlates with viral load. (a) PBMCs were incubated with CM or K562 target cells as per Figure 2. Cells were stained for CD56 and PD-1 expression. NK cell PD-1 expression was assessed against viral load. (b) PBMCs were cultured with 1 µg/mL of β-gal, core, or a HCV protein cocktail of core, NS3 and NS4 or IL-2 (1,000 IU) as indicated. Cells were harvested at 24 h and stained for PD-1 expression. Four replicates from one experiment shown. (c) As per panel a, cells were also stained for CD56, PD-1 and IFN-γ expression. Significant differences are indicated as *P < 0.05. |
Data analyses
Differences in categorical data were determined by Fisher’s exact test. Correlations were determined with the non-parametric Spearman’s rank test. Differences between low viral loads (LVL) and high viral loads (HVL) were evaluated using the non-parametric Mann-Whitney U test. For all tests, P < 0.05 (two-tailed) was considered significant.
| Results | ▴Top |
Subject demographics and infection characteristics
Twenty-three individuals consented to provide blood for this analysis. The majority were Caucasian (66.6 %) and female (56.6%). The median age was 50.5 (range 23 - 65) years. Eighty percent were infected with HCV genotype 1. The median baseline viral load was 1.3 × 106 IU/mL. LVL and HVL were defined a priori as < 800,000 IU/mL (n = 10) and > 800,000 IU/mL (n = 13) respectively, based on previously described associations with therapeutic outcomes for genotype 1 infections [36-38]. Subject demographics and infection characteristics did not differ between the LVL and HVL cohort (Supplementary Table 1, www.gastrores.org).
NK cell cytotoxicity inversely correlates with viral load
Initially, we examined the ability of NK cells derived from HCV infected individuals to lyse K562 target cells with respect to their corresponding viral load. This analysis demonstrated that NK cell cytotoxicity against K562 cells negatively correlated with viral loads (r = -0.437, P = 0.037, Fig. 1a). A similar finding that did not reach statistical significance was observed when NK cells were stimulated with IFN-α prior to K562 exposure (r = -0.372, P = 0.081). When analyzed for dynamics within each viral load cohort, NK cells of the LVL cohort had a greater intrinsic killing capacity, as observed in the presence of medium alone (P < 0.05) or IFN-α (P < 0.05), than that of the HVL cohort (Fig. 1b). Notably, the percentage of NK cells did not differ between patient cohorts (Supplementary Fig. 4, www.gastrores.org).
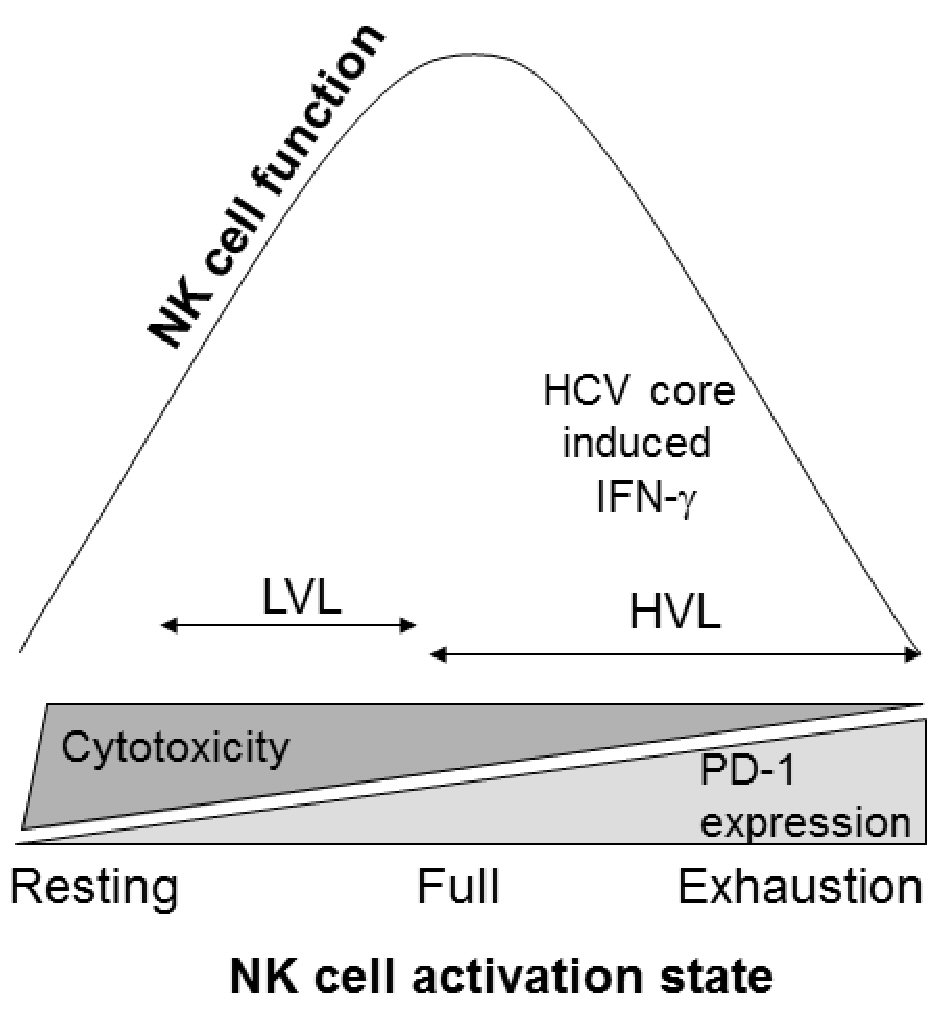 Click for large image | Figure 4. NK cell activation state and function. Cellular activation states move from resting to exhaustion. During LVLs, NK cells appear capable of being further activated allowing for greater NK cell cytotoxicity, along with a capacity for IFN-γ production upon additional stimulation. At HVLs, the constant activation drives the cells towards exhaustion, impairing their ability to function. |
NK cell IFN-γ expression and viral loads
In addition to cytotoxic killing, NK cell IFN-γ expression was evaluated. The percentage of NK cells involved in expressing IFN-γ did not correlate with viral loads (CM, r = 0.334, P = 0.240; K562, r = 0.293, P = 0.290, Fig. 2a) or NK cell cytotoxicity (Supplementary Table 2, www.gastrores.org). The analysis of NK cell IFN-γ expression within the LVL and HVL cohorts, revealed that for the LVL cohort there was a 2.5-fold increase in the percentage of NK cells expressing IFN-γ upon exposure to K562 target cells relative to culture in medium alone (P = 0.0082, Fig. 2b). In contrast, within the HVL cohort, IFN-γ expression was not enhanced by target cells activation.
To investigate whether HCV proteins could directly contribute to cellular IFN-γ synthesis, PBMC were exposed to HCV core protein for 72 h. Unexpectantly, PBMC derived from the LVL cohort were less supportive of HCV core induced IFN-γ synthesis than the HVL cohort (LVL, 1.2 fold HCV core/β-gal; HVL 21.1 fold HCV core/β-gal, P < 0.05, Fig. 2c). Conversely, IL-2 induced IFN-γ synthesis was significantly increased by cells from the LVL cohort (P < 0.05), but not the HVL cohort. A further examination indicated that NK cell IFN-γ expression was directly inducible by the HCV protein cocktail (Supplementary Fig. 2B, www.gastrores.org).
Exhaustion marker PD-1 expression correlates with viral load
The differences in NK responses observed in the LVL and HVL cohorts led us to examine whether NK cells exposed to higher HCV loads were more susceptible to exhaustion, as defined by the appearance of the exhaustion marker PD-1. Flow cytometric assessments of PD-1 expression on NK cells showed that upon K562 stimulation, the percentage of PD-1 NK cells increased proportionally with viral loads (r = 0.649, P < 0.01, Fig. 3a), although NK cell PD-1 expression did not correlate with cytotoxicity (Supplementary Table 2, www.gastrores.org). We also examined whether HCV proteins enhanced NK cell PD-1 expression. Cells from healthy controls and HCV infected individuals were cultured with culture medium, HCV core and a HCV protein cocktail consisting of core, NS3 and NS4. In cells derived from HCV infected individuals, but not healthy individuals, NK cell PD-1 expression was significantly elevated upon overnight exposure to the HCV protein cocktail (P < 0.5, Fig. 3b). The ability of HCV proteins to enhance PD-1 expression and IFN-γ production was supported by the association of PD-1 and IFN-γ expression on NK cells from HCV infected individuals (Fig. 3c).
| Discussion | ▴Top |
HCV infection can alter NK cell function. In this study, we directly investigated the impact of viral load on NK cell function in the clinical setting. It was observed that at LVLs, NK cells had a greater capacity for cytotoxicity than NK cells from a HVL cohort. Moreover, at LVLs, targeted activation of NK cells stimulated increased IFN-γ expression relative to medium alone; whereas, at HVLs IFN-γ expression could not be enhanced beyond baseline levels. In keeping with the lack of responsiveness to stimulation at HVLs, increased viral loads corresponded to greater PD-1 expression. The potential for increased viral loads to affect these events was further supported by the ability of HCV proteins to enhance IFN-γ and PD-1 expression. The potential for IFN-γ and PD-1 to be co-regulated was reinforced by the association between NK cell IFN-γ and PD-1 expression. Taken together these data suggest that under conditions of HVL, HCV protein induced IFN-γ may support PD-1 expression resulting in NK cell exhaustion in chronic HCV infection.
To evaluate changes in NK cell killing with respect to viral load, NK-mediated cytotoxicity was assessed against the prototypic NK cell target K562. A significant negative correlation with viral load was observed (P = 0.037, Fig. 1a). A trend toward similar findings was detected when NK cells were first activated with IFN-α (P = 0.081). The explanation for these findings remains unclear. One possibility is that HCV interfered with NK cell responsiveness to activators such as IFN-α or IL-2 (Fig. 3c) [39, 40]. Thus, at LVLs the ability of NK cells to be stimulated was maintained and as result, cytotoxicity was intact; whereas, at HVLs, HCV interference with NK cell responsiveness reduced the cytotoxic response. Another explanation could be that the extent of stimulation drives NK cell maturation such that less mature NK cells would manifest greater cytotoxic responses than more mature cells [41, 42]. In contrast, NK cell differentiation is associated with an enhanced ability to produce IFN-γ [40]. This is supported by our observation that at LVL NK cells could be stimulated to increase IFN-γ expression, but at HVL, NK cells were already maximally engaged in IFN-γ synthesis (Fig. 2b). The ability of HCV protein to induce IFN-γ could drive this expression with increasing viral loads (Supplementary Fig. 2B, www.gastrores.org). The shift from cytotoxicity to IFN-γ synthesis with increasing viral loads would not only result in ineffective viral control but would also contribute to HCV associated co-morbidities [5, 43]. Although further studies would be required to determine whether in the clinical setting HVLs alter NK cell function or altered NK cell function contributes to HVLs, these processes are likely interdependent. Moreover, even though clear patterns in IFN-γ expression emerged for the LVL and HVL cohorts, the absence of a strict correlation between IFN-γ expression and viral load may reflect physiological nuances due to duration of the infection.
Further supporting reports that HCV inhibits NK cell cytotoxicity [15, 16, 19, 21, 23, 44] were our findings that HVLs were associated with NK cell expression of the exhaustion marker PD-1. CD8 T cell exhaustion is considered key in the progression from acute to chronic HCV infection [32, 45]. Independent studies have observed that CD8 T cell quiescence in HCV infection was linked to the presence of the exhaustion marker PD-1. The effect of PD-1 on T cell cytotoxicity and IFN-γ production has also been elucidated in other viral infection and tumor models [26, 42, 46-48]. However, there is limited information on PD-1 expression and NK cell activity [24, 25, 27, 49]. Our examination of the relationship between NK cell PD-1 expression and viral titer, found that the percentage of NK cells expressing PD-1 increased with viral load (Fig. 3b), suggesting that HCV may upregulate PD-1 expression and thereby suppress NK function. This extends the finding of Golden-Mason et al, that NK cell PD-1 expression from HCV infected individuals was significantly higher in comparison to healthy control populations [24]. This dichotomy in NK PD-1 expression between chronically ill and healthy individuals has also been observed for Epstein-Barr virus (EBV) infections with active lymphomas, tuberculosis and multiple myeloma [25, 27, 49]. To evaluate whether HCV proteins might directly activate PD-1 expression, PBMCs were cultured with HCV proteins core, NS3 and NS4. Relative to healthy cells, the presence of chronic HCV infection was associated with a greater susceptibility to HCV protein induced PD-1 expression (Fig. 3b). The ability of HCV proteins to upregulate PD-1 expression was also observed in the corresponding NKT cell population (Supplementary Fig. 5, www.gastrores.org). In vitro activation of NK cells with Mycobacterium tuberculosis was also shown to initiate PD-1 expression [49]. In keeping with our observations, in HCV infection high viral loads may also upregulate NK cell PD-1 expression on interaction with HCV proteins.
In contrast to cytotoxic responses (Supplementary Table 2, www.gastrores.org), NK cell PD-1 levels were directly proportional to IFN-γ expression (Fig. 3b). The literature suggests a complicated interaction between PD-1 and IFN-γ expression. Reports evaluating the effect of neutralizing the cognate interaction between PD-1 and PD-ligand1 demonstrated enhanced NK cytotoxicity and IFN-γ production in tumor and infection models [25, 26]. Anti-PD-1 antibody, for example, enhanced IFN-γ production against EBV target cells in controlled and active EBV infection (n = 4) [27]. These studies indicate that PD-1 expression results in an impairment of IFN-γ synthesis. However, Alvarez et al observed that in cells isolated from patients with tuberculosis, NK cell PD-1 expression correlated with IFN-γ expression upon activation with M. tuberculosis [49]. Moreover, in the presence of M. tuberculosis the addition of IFN-γ enhanced NK cell PD-1 expression. This is despite their observation that PD-1 blockade with anti-PD-1 or anti-PD-L1 antibody further enhanced IFN-γ synthesis by isolated NK cells [49], as otherwise reported [25-27]. Taken together it appears that with increasing pathogen load IFN-γ synthesis acts as an initiator of PD-1 expression which subsequently serves as negative feedback to keep IFN-γ synthesis in check. The IFN-γ induced upregulation of PD-1 may explain our observed correlation between the percentage of NK cells expressing PD-1 and IFN-γ.
Overall, we propose that NK cell activation in a LVL environment moves from resting toward fully activated cells and in a HVL environment from fully activated towards exhaustion (Fig. 4). Thus, NK cell cytotoxicity is the strongest under LVL conditions and diminishes in HVL conditions as cells become exhausted. This cellular progression from fully active to exhaustion appears to be supported by HCV protein, but not IL-2, induced NK cell IFN-γ expression in the HVL cohort (Fig. 3c). As result, in the setting of HVLs, NK cells may be terminally differentiated for IFN-γ synthesis, preventing responsiveness to further stimulation (Fig. 2b). It may be this condition, which allows for IFN-γ upregulation of exhaustion marker PD-1 expression at HVLs.
This study has a number of caveats. First, the sample size was small which could contribute to type I or type II statistical errors [27]. Second, peripheral NK cell function was not examined during treatment or at the end of treatment to evaluate how NK cell activity changed due to the effect of treatment on viral load. Finally, the duration of infection was unknown. Therefore, it could not be determined whether NK cell exhaustion reflects the viral load or the duration of infection. Thus, in addition to temporal changes in the number and function of NK cells over the course of HCV infection, further studies are also needed to determine whether NK cell exhaustion is related to viral load or the duration of infection.
In conclusion, our study suggests that with increasing viral loads, NK cells become further differentiated resulting in reduced cytotoxicity and enhanced IFN-γ production. This state is associated with enhanced NK cell PD-1 expression at HVLs. Thus, these data indicate that under conditions of HVL, ineffective NK cell viral control is linked to exhaustion in chronic HCV infection.
Acknowledgments
The authors wish to thank Ms. R. Vizniak for her prompt and accurate typing of the manuscript.
Grant Support
This research was funded by a grant from the Canadian Liver Foundation and the Morberg Family Chair in Hepatology.
Conflict of Interest
The authors have no conflicts of interest to disclose.
Abbreviations
HCV: hepatitis C virus; NK: natural killer; IFN: interferon; PBMC: peripheral blood mononuclear cell; cpm: counts per minute; ICCS: intracellular cytokine staining; EBV: Epstein-Barr virus; LVL: low viral load; HVL: high viral load
| References | ▴Top |
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5(9):558-567.
doi - Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002;36(5 Suppl 1):S21-29.
pubmed - Dolganiuc A, Chang S, Kodys K, Mandrekar P, Bakis G, Cormier M, Szabo G. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol. 2006;177(10):6758-6768.
doi pubmed - Heim MH, Thimme R. Innate and adaptive immune responses in HCV infections. J Hepatol. 2014;61(1 Suppl):S14-25.
doi pubmed - Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. 2013;19(7):859-868.
doi pubmed - Holder KA, Russell RS, Grant MD. Natural killer cell function and dysfunction in hepatitis C virus infection. Biomed Res Int. 2014;2014:903764.
doi pubmed - Abdel-Hakeem MS, Shoukry NH. Protective immunity against hepatitis C: many shades of gray. Front Immunol. 2014;5:274.
doi - Golden-Mason L, Rosen HR. Natural killer cells: multifaceted players with key roles in hepatitis C immunity. Immunol Rev. 2013;255(1):68-81.
doi pubmed - Moretta A, Bottino C, Mingari MC, Biassoni R, Moretta L. What is a natural killer cell? Nat Immunol. 2002;3(1):6-8.
doi pubmed - Raulet DH. Interplay of natural killer cells and their receptors with the adaptive immune response. Nat Immunol. 2004;5(10):996-1002.
doi pubmed - Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187-376.
doi - Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189-220.
doi pubmed - Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, Moretta L. Receptors for HLA class-I molecules in human natural killer cells. Annu Rev Immunol. 1996;14:619-648.
doi pubmed - Shoukry NH, Pelletier S, Chang KM. A view to natural killer cells in hepatitis C. Gastroenterology. 2011;141(4):1144-1148.
doi pubmed - Crotta S, Stilla A, Wack A, D'Andrea A, Nuti S, D'Oro U, Mosca M, et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med. 2002;195(1):35-41.
doi pubmed - Tseng CT, Klimpel GR. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J Exp Med. 2002;195(1):43-49.
doi - Lau DT, Negash A, Chen J, Crochet N, Sinha M, Zhang Y, Guedj J, et al. Innate immune tolerance and the role of kupffer cells in differential responses to interferon therapy among patients with HCV genotype 1 infection. Gastroenterology. 2013;144(2):402-413 e412.
- Morishima C, Paschal DM, Wang CC, Yoshihara CS, Wood BL, Yeo AE, Emerson SS, et al. Decreased NK cell frequency in chronic hepatitis C does not affect ex vivo cytolytic killing. Hepatology. 2006;43(3):573-580.
doi pubmed - Dessouki O, Kamiya Y, Nagahama H, Tanaka M, Suzu S, Sasaki Y, Okada S. Chronic hepatitis C viral infection reduces NK cell frequency and suppresses cytokine secretion: Reversion by anti-viral treatment. Biochem Biophys Res Commun. 2010;393(2):331-337.
doi pubmed - Bonorino P, Ramzan M, Camous X, Dufeu-Duchesne T, Thelu MA, Sturm N, Dariz A, et al. Fine characterization of intrahepatic NK cells expressing natural killer receptors in chronic hepatitis B and C. J Hepatol. 2009;51(3):458-467.
doi pubmed - Nattermann J, Feldmann G, Ahlenstiel G, Langhans B, Sauerbruch T, Spengler U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut. 2006;55(6):869-877.
doi pubmed - Corado J, Toro F, Rivera H, Bianco NE, Deibis L, De Sanctis JB. Impairment of natural killer (NK) cytotoxic activity in hepatitis C virus (HCV) infection. Clin Exp Immunol. 1997;109(3):451-457.
doi pubmed - Kawarabayashi N, Seki S, Hatsuse K, Ohkawa T, Koike Y, Aihara T, Habu Y, et al. Decrease of CD56(+)T cells and natural killer cells in cirrhotic livers with hepatitis C may be involved in their susceptibility to hepatocellular carcinoma. Hepatology. 2000;32(5):962-969.
doi pubmed - Golden-Mason L, Klarquist J, Wahed AS, Rosen HR. Cutting edge: programmed death-1 expression is increased on immunocytes in chronic hepatitis C virus and predicts failure of response to antiviral therapy: race-dependent differences. J Immunol. 2008;180(6):3637-3641.
doi pubmed - Benson DM, Jr., Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, Baiocchi RA, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116(13):2286-2294.
doi pubmed - Dolina JS, Sung SS, Novobrantseva TI, Nguyen TM, Hahn YS. Lipidoid nanoparticles containing PD-L1 siRNA delivered in vivo enter kupffer cells and enhance NK and CD8(+) T cell-mediated hepatic antiviral immunity. Mol Ther Nucleic Acids. 2013;2:e72.
doi pubmed - Wiesmayr S, Webber SA, Macedo C, Popescu I, Smith L, Luce J, Metes D. Decreased NKp46 and NKG2D and elevated PD-1 are associated with altered NK-cell function in pediatric transplant patients with PTLD. Eur J Immunol. 2012;42(2):541-550.
doi pubmed - Kamphorst AO, Ahmed R. Manipulating the PD-1 pathway to improve immunity. Curr Opin Immunol. 2013;25(3):381-388.
doi pubmed - Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol. 2013;6(1):74.
doi pubmed - Porichis F, Hart MG, Zupkosky J, Barblu L, Kwon DS, McMullen A, Brennan T, et al. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J Virol. 2014;88(5):2508-2518.
doi pubmed - Sumida K, Shimoda S, Iwasaka S, Hisamoto S, Kawanaka H, Akahoshi T, Ikegami T, et al. Characteristics of splenic CD8+ T cell exhaustion in patients with hepatitis C. Clin Exp Immunol. 2013;174(1):172-178.
doi pubmed - Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. 2007;81(17):9249-9258.
doi pubmed - Aborsangaya KB, Dembinski I, Khatkar S, Alphonse MP, Nickerson P, Rempel JD. Impact of aboriginal ethnicity on HCV core-induced IL-10 synthesis: interaction with IL-10 gene polymorphisms. Hepatology. 2007;45(3):623-630.
doi pubmed - Tran J, Kung SK. Lentiviral vectors mediate stable and efficient gene delivery into primary murine natural killer cells. Mol Ther. 2007;15(7):1331-1339.
doi pubmed - Rempel JD, Packiasamy J, Dean HJ, McGavock J, Janke A, Collister M, Wicklow B, et al. Preliminary analysis of immune activation in early onset type 2 diabetes. Int J Circumpolar Health. 2013;72(1):21190.
doi - Martinot-Peignoux M, Marcellin P, Pouteau M, Castelnau C, Boyer N, Poliquin M, Degott C, et al. Pretreatment serum hepatitis C virus RNA levels and hepatitis C virus genotype are the main and independent prognostic factors of sustained response to interferon alfa therapy in chronic hepatitis C. Hepatology. 1995;22(4 Pt 1):1050-1056.
doi pubmed - Martinot-Peignoux M, Boyer N, Pouteau M, Castelnau C, Giuily N, Duchatelle V, Auperin A, et al. Predictors of sustained response to alpha interferon therapy in chronic hepatitis C. J Hepatol. 1998;29(2):214-223.
doi - Berg T, Sarrazin C, Herrmann E, Hinrichsen H, Gerlach T, Zachoval R, Wiedenmann B, et al. Prediction of treatment outcome in patients with chronic hepatitis C: significance of baseline parameters and viral dynamics during therapy. Hepatology. 2003;37(3):600-609.
doi pubmed - Rempel JD, Aborsangaya KB, Alphonse MP, Minuk GY. The influence of North American Aboriginal ethnicity on pro-inflammatory and anti-inflammatory cytokine responses to IFN-alpha. J Viral Hepat. 2009;16(4):292-297.
doi pubmed - Luetke-Eversloh M, Cicek BB, Siracusa F, Thom JT, Hamann A, Frischbutter S, Baumgrass R, et al. NK cells gain higher IFN-gamma competence during terminal differentiation. Eur J Immunol. 2014;44(7):2074-2084.
doi pubmed - Vahlne G, Becker S, Brodin P, Johansson MH. IFN-gamma production and degranulation are differentially regulated in response to stimulation in murine natural killer cells. Scand J Immunol. 2008;67(1):1-11.
pubmed - Yu P, Steel JC, Zhang M, Morris JC, Waldmann TA. Simultaneous blockade of multiple immune system inhibitory checkpoints enhances antitumor activity mediated by interleukin-15 in a murine metastatic colon carcinoma model. Clin Cancer Res. 2010;16(24):6019-6028.
doi pubmed - Cacoub P, Gragnani L, Comarmond C, Zignego AL. Extrahepatic manifestations of chronic hepatitis C virus infection. Dig Liver Dis. 2014;46(Suppl 5):S165-173.
doi pubmed - Holder KA, Stapleton SN, Gallant ME, Russell RS, Grant MD. Hepatitis C virus-infected cells downregulate NKp30 and inhibit ex vivo NK cell functions. J Immunol. 2013;191(6):3308-3318.
doi pubmed - Thimme R, Bukh J, Spangenberg HC, Wieland S, Pemberton J, Steiger C, Govindarajan S, et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci U S A. 2002;99(24):15661-15668.
doi pubmed - Hallett WH, Jing W, Drobyski WR, Johnson BD. Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biol Blood Marrow Transplant. 2011;17(8):1133-1145.
doi pubmed - Okazaki T, Honjo T. Rejuvenating exhausted T cells during chronic viral infection. Cell. 2006;124(3):459-461.
doi pubmed - Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682-687.
doi pubmed - Alvarez IB, Pasquinelli V, Jurado JO, Abbate E, Musella RM, de la Barrera SS, Garcia VE. Role played by the programmed death-1-programmed death ligand pathway during innate immunity against Mycobacterium tuberculosis. J Infect Dis. 2010;202(4):524-532.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.