

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website http://www.gastrores.org |
Case Report
Volume 11, Number 2, April 2018, pages 150-153
Peutz-Jeghers Syndrome Presenting as Colonic Intussusception: A Rare Entity
Jamil Shaha, Tagore Sunkaraa, e, Philip Xiaob, Vinaya Gaduputic, Madhavi Reddya, Sultana Raziad
aDivision of Gastroenterology & Hepatology, The Brooklyn Hospital Center, 121 Dekalb Ave, Brooklyn, NY 11201, USA
bDivision of Pathiology, The Brooklyn Hospital Center, 121 Dekalb Ave, Brooklyn, NY 11201, USA
cDivision of Gastroenterology & Hepatology, SBH Health System, 4422 Third Ave, Bronx, NY 10457, USA
dDivision of Gastroenterology, Slocum-Dickson Medical Group, 1729 Burrstone Road, New Hartford, NY 13413, USA
eCorresponding Author: Tagore Sunkara, The Brooklyn Hospital Center, Clinical Affiliate of Mount Sinai Hospital, 121 Dekalb Avenue, Brooklyn, NY 11201, USA
Manuscript submitted December 22, 2017, accepted February 28, 2018
Short title: Peutz-Jeghers Syndrome as Colonic Intussusception
doi: https://doi.org/10.14740/gr964e
| Abstract | ▴Top |
Peutz-Jeghers syndrome is an autosomal dominant inherited medical condition characterized by hyperpigmented mucocutaneous macules, hamartomatous polyps in the digestive tract, and with a greater risk of gastrointestinal and non-gastrointestinal cancers. In fact, without appropriate medical surveillance, the lifetime risk for all cancers combined may be as high as 93%. The syndrome is rare, with estimates of incidence varying between 1 in 8,300 and 1 in 280,000 live births. Infrequently, individuals present for the first time with bowel obstruction secondary to intussusception. Here, we present an interesting case of a young Burmese man who, early on, showed traits of Peutz-Jeghers syndrome, including the characteristic hyperpigmented areas on the fingers and lips. Unfortunately, the diagnosis was not made until he later developed bowel obstruction caused by an intussusception, requiring exploratory laparoscopic bowel resection. A high index of suspicion is needed to diagnose accurately. However, early identification and close surveillance can lead to excellent prognosis in these individuals.
Keywords: Peutz-Jeghers syndrome; Colon polyps; Intussusception; Colon cancer; Hamartomatous polyps; Bowel obstruction
| Introduction | ▴Top |
Peutz-Jeghers syndrome is an autosomal dominant inherited medical condition characterized by hamartomatous polyps in the digestive tract as well as hyperpigmented mucocutaneous macules on the lips, mucous membranes of the mouth, hands, and feet. Although the hamartomatous polyps are non-cancerous growths with low malignant potential, individuals with this syndrome are at elevated risk of acquiring malignancies of the breast, colorectal tract, pancreas, stomach, small intestine, liver, lung, testicles, ovaries, cervix, uterus, and other organs [1, 2]. The syndrome is rare, with estimates of incidence varying between 1 in 8,300 and 1 in 280,000 live births [3]. Here, we present a rare case of a young man with Peutz-Jeghers syndrome.
| Case Report | ▴Top |
An 18-year-old Burmese man, with no significant medical history at the time, visited the Emergency Department for intermittent abdominal pain that had acutely worsened. He reported no fevers, shortness of breath, lightheadedness, fatigue, blood in the stool, tarry stools, changes in bowel habits, weight loss, nor any other symptoms other than severe abdominal pain. His vital signs were normal. Physical examination was remarkable for left upper quadrant abdominal tenderness and mild abdominal distention. Also, small, dark blue-colored spots on the lips (Fig. 1) as well as the fingers were noted (Fig. 2). Laboratory testing was notable for hemoglobin of 8.3 g/dL with an unknown baseline. He had a mean corpuscular volume of 74 fL, a red blood cell distribution width of 17.2%, as well as an iron panel with iron level of 18 µg/dL, iron-binding capacity of 362 µg/dL, and ferritin of 35 µg/dL findings consistent with iron-deficiency anemia.
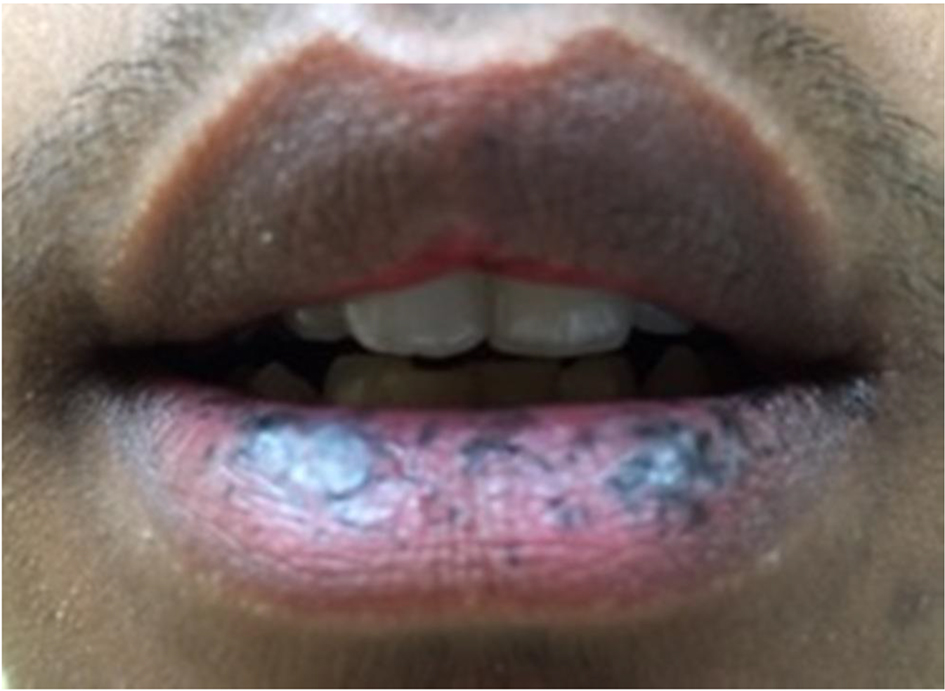 Click for large image | Figure 1. Characteristic hyperpigmented macules of Peutz-Jeghers syndrome on the patient’s lips. |
 Click for large image | Figure 2. Characteristic hyperpigmented macules of Peutz-Jeghers syndrome on the patient’s fingers. |
Abdominal computerized tomography (CT) scan was performed and showed findings of a segment of large intestine invaginating into the adjoining intestinal lumen at the level of the splenic flexure (Fig. 3). The patient was taken to the operating room where he underwent exploratory laparoscopic bowel resection in order to surgically treat an intussusception that had caused large bowel obstruction. He was found to have multiple large polyps of varying sizes which were sent for histopathological evaluation. The pathology report showed hamartomatous polyps consistent with Peutz-Jeghers polyposis. There was no malignancy and the margins were negative. The patient was started on oral iron supplementation and referred to a gastroenterologist for further evaluation. He underwent colonoscopy where he was found to have many polyps throughout the colon and rectum (Fig. 4), the largest of which were removed and sent for histopathological evaluation. The histology revealed a polyp with proliferation of the smooth muscle bundle (Fig. 5a) on hematoxylin and eosin stain (× 400). Immunohistochemistry (IHC) stained positive for smooth muscle actin (SMA) (× 100) indicating smooth muscle origin with central core branching of the smooth muscle (Fig. 5b) consistent with hamartomatous polyps. Also, the patient underwent genetic testing, which was positive for a germline mutation in the STK11 (LKB1) gene, confirming the diagnosis of Peutz-Jeghers syndrome. His family members have since been referred for genetic testing and colonoscopy as they none of the family members had a colonoscopy done.
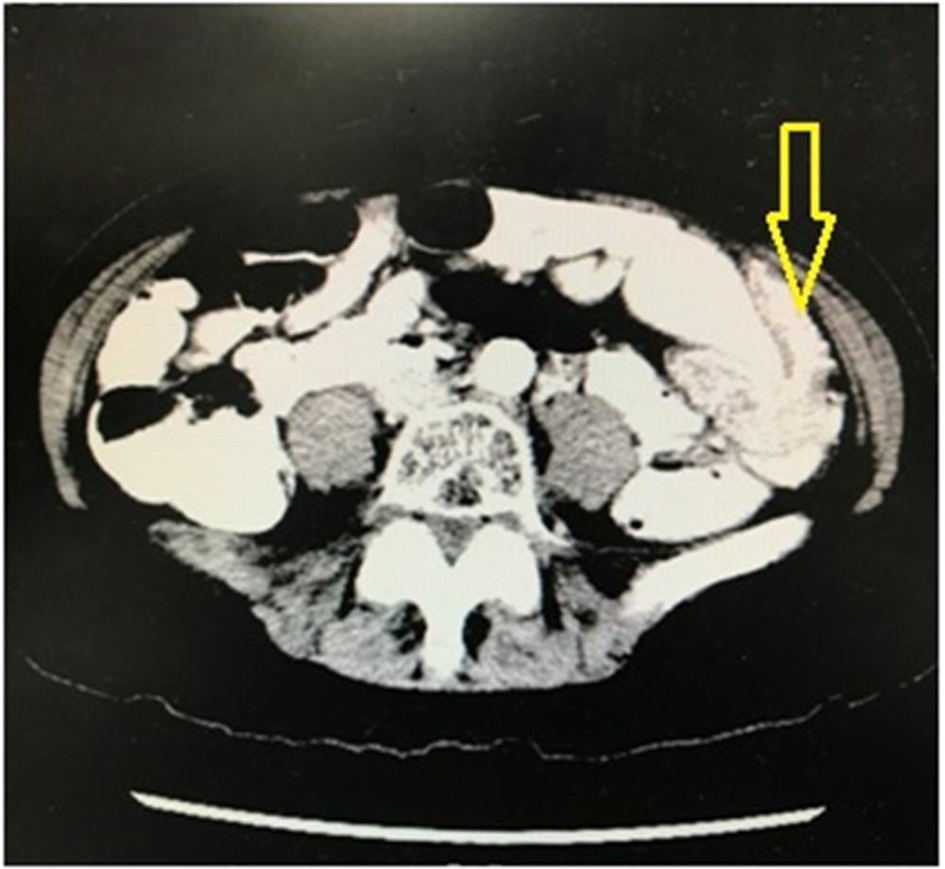 Click for large image | Figure 3. Computerized tomography (CT) scan of the abdomen showing findings of a segment of large intestine invaginating into the adjoining intestinal lumen at the level of the splenic flexure, consistent with an intussusception that had caused large bowel obstruction. |
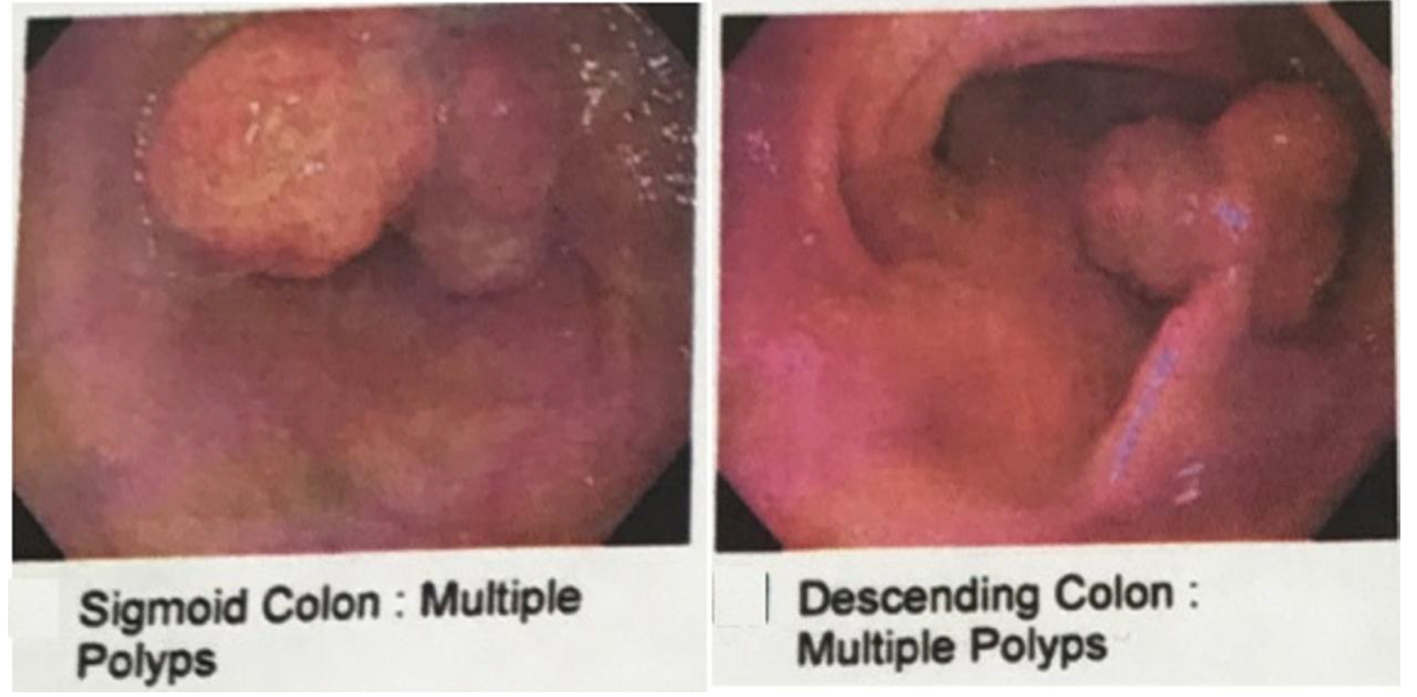 Click for large image | Figure 4. Colonoscopy showing multiple polyps in the descending colon and sigmoid colon. |
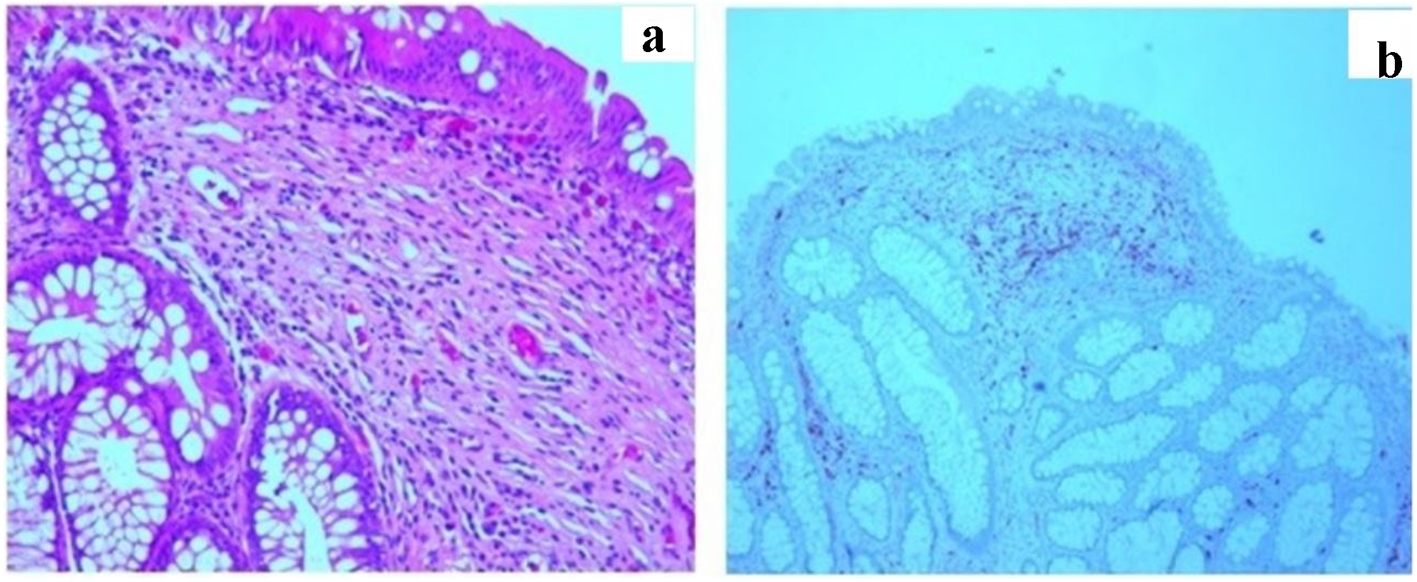 Click for large image | Figure 5. (a) Biopsies (hematoxylin and eosin stain) (× 400) revealed a polyp with proliferation of the smooth muscle bundle. (b) Immunohistochemistry (IHC) stained positive for smooth muscle actin (SMA) (× 100) indicating smooth muscle origin with central core branching of the smooth muscle consistent with hamartomatous polyps. |
| Discussion | ▴Top |
Intussusception takes place when a loop of bowel, the intussusceptum, telescopes into an adjoining segment, the intussuscipiens. This clinical presentation can be seen in both young and adult individuals with Peutz-Jeghers syndrome and may be the initial presentation for some of them. Most cases of intussusceptions in Peutz-Jeghers syndrome are due to polyps located in the small intestine [4]. The majority that have been reported in the literature are either ileal or jejunal [5]. The duodenum is an uncommon location for intussusception, as it lies in a fixed retroperitoneal position [5]. Colo-colonic intussusception, such as this one, is very rare and has only been reported only in a few cases [6]. Recently, there has been a reported case of colonic intussusception from benign etiology and with no obvious pathology [7].
Peutz-Jeghers syndrome is an autosomal dominant inherited medical condition characterized by hyperpigmented mucocutaneous macules on the lips, mucous membranes of the mouth, hands, and feet, as well as hamartomatous polyps in the digestive tract. These polyps are noncancerous growths with low malignant potential. In 1921, Peutz-Jeghers syndrome was initially described in a published case report by Jan Peutz, a Dutch medical doctor who noticed a link between the mucocutaneous macules and the gastrointestinal polyps in members of a Dutch family [8]. It was later formalized into a syndrome in 1949 by Harold Jeghers, an American medical doctor now given credit for the first conclusive informative reports of the syndrome [9].
According to diagnostic criteria of the World Health Organization, a diagnosis of the syndrome should be considered in an individual who satisfies any one of the following: 1) three or more Peutz-Jeghers polyps confirmed by histology; or 2) any number of Peutz-Jeghers polyps with a family history of the syndrome; or 3) typical visible mucocutaneous pigmentation with a family history of the syndrome; or 4) any number of Peutz-Jeghers polyps and typical visible mucocutaneous pigmentation. Individuals who meet clinical criteria may submit to genetic testing for germline mutations in the STK11 (LKB1) gene, a possible tumor suppressor gene on chromosome 19, in order to confirm the diagnosis [2, 10]. Given the autosomal dominant inheritance pattern, family members should be evaluated for diagnostic criteria for Peutz-Jeghers syndrome as well as undergo genetic testing and be counseled if discovered to be positive [10].
There are no definitive consensus guidelines for the management of Peutz-Jeghers polyps, with each case requiring individualized management. In general, if significant bleeding or intussusception occurs, polyp resection is needed [11]. Colonoscopy should be performed to snare polyps, particularly large, single polyps that are within reach, and enterotomy should be performed to surgically resect short lengths of heavily involved intestinal segments [11].
Individuals with Peutz-Jeghers syndrome are at elevated risk of acquiring malignancies of the breast, colorectal tract, pancreas, stomach, small intestine, liver, lung, testicles, ovaries, cervix, uterus, and other organs [1, 2]. In fact, without appropriate medical surveillance, the lifetime risk for all cancers combined may be as high as 93% [12]. As a group, gastrointestinal cancers are the most common malignancies in individuals with Peutz-Jeghers syndrome (63% by age 70) [13]. By comparison, in the overall population, the risk of any gastrointestinal malignancy by age 70 is 18% [3].
Surveillance protocols for individuals with Peutz-Jeghers syndrome have two key objectives. As discussed, one is to identify sizeable intestinal polyps that could lead to intussusception and obstruction or bleeding and anemia. The other is to identify malignancy at an early stage. The screening indications are therefore dependent upon age: polyp-related complications may develop as early as childhood while cancer risk relates mainly to the adult population. Surveillance guidelines include upper endoscopy and colonoscopy (or video capsule endoscopy), starting at 8 years of age [1, 10]. If polyps are detected, the procedure should be performed again every 2 - 3 years [1, 10]. If no polyps are detected, the procedure should be performed again by 18 years of age, and then every 2 - 3 years thereafter [1, 10]. Furthermore, endoscopic ultrasound and/or pancreatic MRI should be performed in individuals with Peutz-Jeghers syndrome to screen for pancreatic cancer, starting at 30 years of age and performed again every 1 - 2 years thereafter [1, 10].
Cancer screening guidelines for individuals with Peutz-Jeghers syndrome are dependent upon gender, as well. Cancer screening for women with Peutz-Jeghers syndrome includes breast self-exam on a monthly basis and clinical breast exam on a yearly basis performed by a medical professional, starting at 20 years of age [1, 10]. Also, mammogram should be performed every 2-3 years, starting at 20 years of age, and then yearly, starting at 40 years of age [1, 10]. Yearly gynecologic exam, including a Pap smear, transvaginal ultrasound, and consideration of a uterine biopsy, should be performed starting at 25 years of age [1, 10]. Cancer screening for men with Peutz-Jeghers syndrome includes testicular exam on a yearly basis, starting in childhood [1, 10]. Testicular ultrasound should be performed to look for abnormalities [1, 10]. Also, any signs of potential hormonal problems should be investigated [1, 10].
Conclusions
Peutz-Jeghers syndrome is an autosomal dominant inherited medical condition. The syndrome is rare and individuals often remain undiagnosed for many years. A high index of suspicion is needed to diagnose accurately. Otherwise, individuals may present for the first time with acute complications such as bowel obstruction secondary to intussusception. However, early identification, in patients and family members, as well as close cancer surveillance can lead to excellent prognosis in these individuals.
Disclosures
None.
Author Contributions
All authors have made contributions to the article and have reviewed it before submission.
Consent
Informed consent for participation was obtained from this patient.
Conflict of Interest
None of the authors have any financial conflict of interest.
| References | ▴Top |
- van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105(6):1258-1264; author reply 1265.
doi pubmed - Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, Bertario L, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975-986.
doi pubmed - Lindor NM, McMaster ML, Lindor CJ, Greene MH. Concise handbook of familial cancer susceptibility syndromes - second edition. J Natl Cancer Inst Monogr. 2008;38:1-93.
doi - Wang H, Luo T, Liu WQ, Huang Y, Wu XT, Wang XJ. Clinical presentations and surgical approach of acute intussusception caused by Peutz-Jeghers syndrome in adults. J Gastrointest Surg. 2011;15(12):2218-2225.
doi pubmed - Chen XD, Yu YY, Yang L, Rui YY, Zhou ZG. Duodenal intussusception due to a giant neuroendocrine carcinoma in a patient with Peutz-Jeghers syndrome: case report and systematic review. Eur J Gastroenterol Hepatol. 2012;24(6):722-726.
doi pubmed - Jaremko JL, Rawat B. Colo-colonic intussusception caused by a solitary Peutz-Jeghers polyp. Br J Radiol. 2005;78(935):1047-1049.
doi pubmed - Sunkara T, Caughey M E, Culliford A, Gaduputi V. A Rare Case of Adult Colonic Intussusception from Benign Etiology. Cureus.2018;10(1):e2023.
doi - Peutz JL. Over een zeer merkwaardige, gecombineerde familiaire pollyposis van de sligmliezen van den tractus intestinalis met die van de neuskeelholte en gepaard met eigenaardige pigmentaties van huid-en slijmvliezen. Ned Maandschr v Gen. 1921;10:134.
- Jeghers H, Mc KV, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241(26):1031-1036.
doi pubmed - Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW, American College of G. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223-262; quiz 263.
doi pubmed - Duan SX, Wang GH, Zhong J, Ou WH, Fu MX, Wang FS, Ma SH, et al. Peutz-Jeghers syndrome with intermittent upper intestinal obstruction: A case report and review of the literature. Medicine (Baltimore). 2017;96(17):e6538.
doi pubmed - Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, Cruz-Correa M, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447-1453.
doi pubmed - Mehenni H, Resta N, Park JG, Miyaki M, Guanti G, Costanza MC. Cancer risks in LKB1 germline mutation carriers. Gut. 2006;55(7):984-990.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.