










| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website http://www.gastrores.org |
Case Report
Volume 11, Number 1, February 2018, pages 64-67
A Case of Adolescent Cronkhite-Canada Syndrome
Mikaell Alexandre Gouvea Fariaa, b, d, Bruna Basagliab, Vinicius Quintiliano Moutinho Nogueiraa, Tatiana Barros Gama Ferraz de Mendoncaa, Roberto Luiz Kaiser Juniora, Idiberto Jose Zotarelli Filhoa, c, Luiz Gustavo de Quadrosa
aKaiser Clinic and Hospital, Sao Jose do Rio Preto, SP Brazil
bUniao Das Faculdades Dos Grandes Lagos (Unilago) Medical School, Sao Jose do Rio Preto, SP, Brazil
cSao Paulo State University (Unesp), Institute of Biosciences, Humanities and Exact Sciences (Ibilce), Campus Sao Jose do Rio Preto, SP, Brazil
dCorresponding Author: Mikaell Alexandre Gouvea Faria, Division of Gastroenterology of Medical School (Unilago) and Kaiser Clinic and Hospital, Sao Jose do Rio Preto, SP, Brazil.
Manuscript submitted September 22, 2017, accepted October 9, 2017
Short title: An Adolescent Cronkhite-Canada Syndrome Case
doi: https://doi.org/10.14740/gr912w
| Abstract | ▴Top |
The Cronkhite-Canada syndrome (CCS) was first reported in 1955, since then more than 500 cases have been reported, indicating an estimated incidence of one case per 1 million inhabitants. The syndrome occurs predominantly in male, with a ratio of three males to two females. Because of the rarity of the syndrome, there are no evidence-based therapies and the treatments described include a combination of therapies, but none is consistently effective. Surgery is usually reserved for the treatment of complications. Herein, we present a case of adolescent CCS. The patient was a 15-year-old boy who presented with watery diarrhea with 20 episodes a day, vomiting and abdominal pain for 4 weeks, with a weight loss of 8.0 kg (15.0% of initial weight). Endoscopic examination revealed polyposis in the stomach, duodenum, and colon. CCS was diagnosed and the patient was treated with a combined corticosteroid and metronidazole. Followed up at 8 month after the diagnosis, the patient was asymptomatic.
Keywords: Cronkhite-Canada syndrome; Symptoms; Treatment; Rare case
| Introduction | ▴Top |
The Cronkhite-Canada syndrome (CCS) was first reported in 1955, since then more than 500 cases have been reported [1, 2], indicating an estimated incidence of one case per 1 million inhabitants. The reported cases of Cronkhite-Canada infant syndrome are scarce (< 10) [3].
There is no data available on racial predisposition, with a worldwide distribution [3], but around 75.0% of reported cases are from Japan [4]. CCS affects predominantly male, with a ratio of three males to two females [3].
Most patients are older than 50 years and have the symptoms of diarrhea, weight loss, loss of appetite, abdominal pain and weakness. With regard to cutaneous symptoms, they occur as a result of intestinal malabsorption [3]. Because of the rarity of the syndrome, there are no evidence-based therapies and the treatments described include a combination of therapies, but none is consistently effective. Surgery is usually reserved for the treatment of complications [2, 5].
The objective of the present study was to analyze and establish the best clinical and drug treatment (combined therapy) for the present case of CCS belonging to Kaiser Clinic of Sao Jose do Rio Preto, Sao Paulo, Brazil. Through the present case report of CCS, an extremely rare clinical entity, it will correlate with the findings and treatments of the present case with the other reports in the literature, through a comprehensive review of medical literature in order to highlight and provide information on this topic.
| Case Report | ▴Top |
A 15-year-old male presented with watery diarrhea with 20 episodes a day, vomiting and abdominal pain for 4 weeks, with a weight loss of 8 kg (15.0% of initial weight). He also had brownish spots on his hands, lateral surface of the cocks and feet, hyperchromic punctate lesions on the trunk and onychodystrophy. His abnormal pain was often worsened by ingestion of spicy foods and pepper. The patient had a history of cleft lip and palate and reported recurrent diarrheal episodes since childhood.
Laboratory findings included hemoglobin 15.1 g/dL (11.5 - 14.8), hematocrit 49.0% (40-50%), discrete anisocytosis and hypochromia; total leukocyte number 7,500/mm3 (4,500 - 11,000), with platelets 167,000/mm3 (150,000 - 350,000); fasting glycemia 67.0 mg/dL (70.0 - 99.0), creatinine 0.73 mg/dL (0.70 - 1.20), potassium 4.0 mmol/L (3.5 - 5.5), calcium 8.7 mg/dL (8.9 - 10.1), magnesium 1.9 mg/dL (1.7 - 2.1), zinc 51.0 µg/dL (70.0 - 120.0); total proteins 3.52 g/dL (6.6 - 8.3), albumin 2.09 g/dL (3.5 - 5.2) and globulins 1.43 g/dL (1.8 - 4.4); serum iron 149.0 µg/dL (70.0 - 180.0), ferritin 130.49 ng/dL (30.0 - 200.0), 25-hydroxy vitamin D 36.95 ng/mL (30.00 - 100.00), vitamin B12 below 150.0 pg/mL (300.0 - 900.0); total bilirubin 0.94 mg/dL (0.30 - 1.90), (direct 0.16 mg/dL and indirect 0.78 mg/dL), AST 24.0 IU/L (until 37.0), alkaline phosphatase 172.0 IU/L (74.0 - 390.0), gamma-GT 22.0 IU/L (7.0 - 60.0); Total cholesterol 183.0 mg/dL (< 170.0), HDL 43.0 mg/dL (> 45.0), LDL 118.0 mg/dL (< 110.0), VLDL 22.0 mg/dL, triglycerides 110.0 mg/dL (< 90.0); TSH 1.79 µIU/mL (0.40 - 4.50), free T4 1.04 ng/dL (0.7 - 1.8), total testosterone 710.78 ng/dL (241.00 - 827.00), and free testosterone 14,614 ng/dL(undefined).
Esophagogastroduodenoscopy revealed the presence of multiple types II and III polyps of the Yamada morphological classification on areas of atrophic mucosa throughout the stomach, and multiple nodular lymphoid lesions in bulb and second duodenal portion with atrophic mucosa (Fig. 1). Colonoscopy revealed multiple sessile and subpediculated polyps on normal-appearing mucosa, throughout the colon and rectum (Fig. 2).
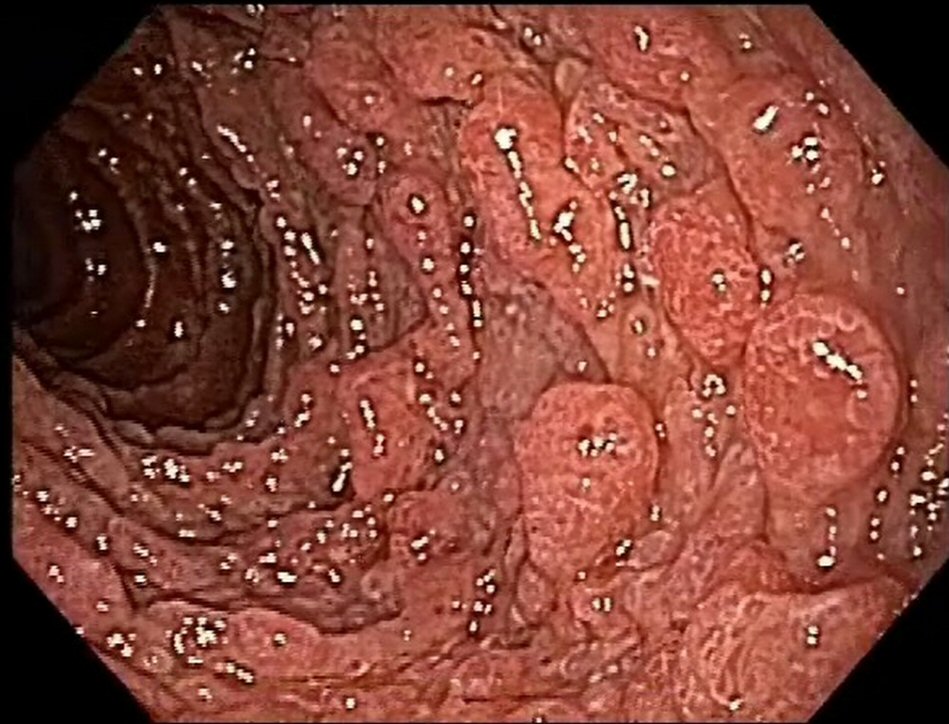 Click for large image | Figure 1. Upper endoscopy reveals polyposis in duodenum. |
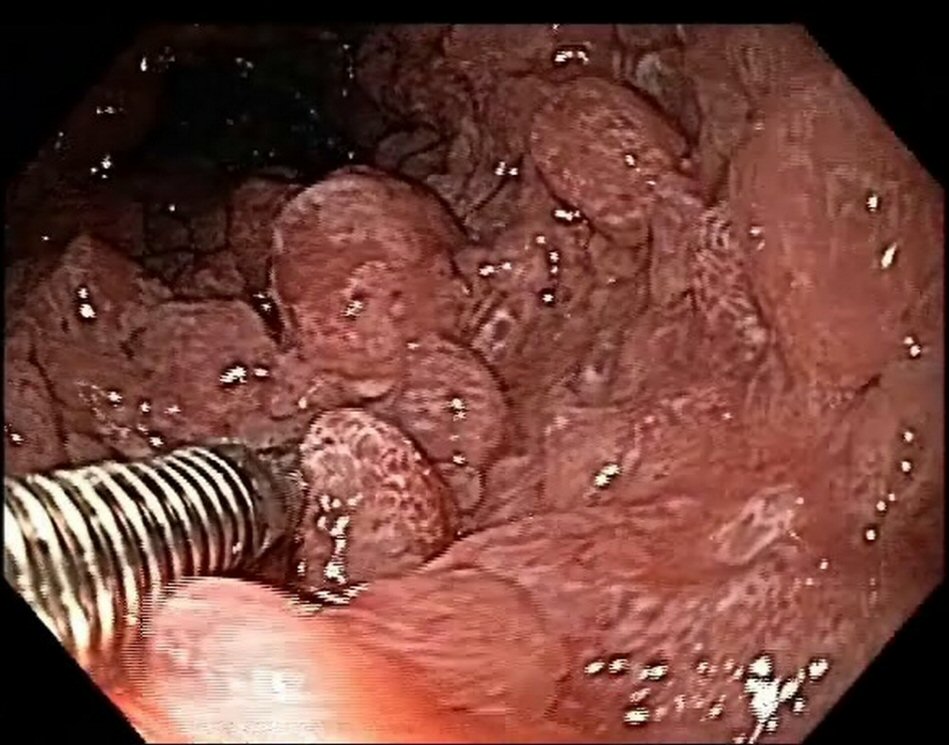 Click for large image | Figure 2. Colonoscopy reveals polyposis. |
Based on these findings, the diagnosis of CCS was suggested, treatment with a systemic corticosteroid at a dose of 40 mg/day of prednisone for 60 days was started, associated with the use of metronidazole 1,200 mg/day for 7 days. The patient progressed with gradual improvement of symptoms and weight gain, and weaned slowly from corticosteroid therapy until its complete suspension in 160 days, when it reached a weight of 58 kg and complete resolution of the symptoms. Diarrhea and cutaneous condition recurred 5 days after the suspension of the therapy, necessitating the reintroduction of prednisone at dose of 20.0 mg/day, with symptomatic remission.
The patient was doing well 9 months after the onset of the condition; there were two bowel movements per day, with solid stools and weight gain, with rare reports of diarrheal episodes using seasoned foods. The patient also underwent follow-up esophagogastroduodenoscopy and colonoscopy 8 months after the diagnosis. Despite the clinical remission, there was endoscopic evidence of polyposis (Fig. 3, 4). This case did not show anemia.
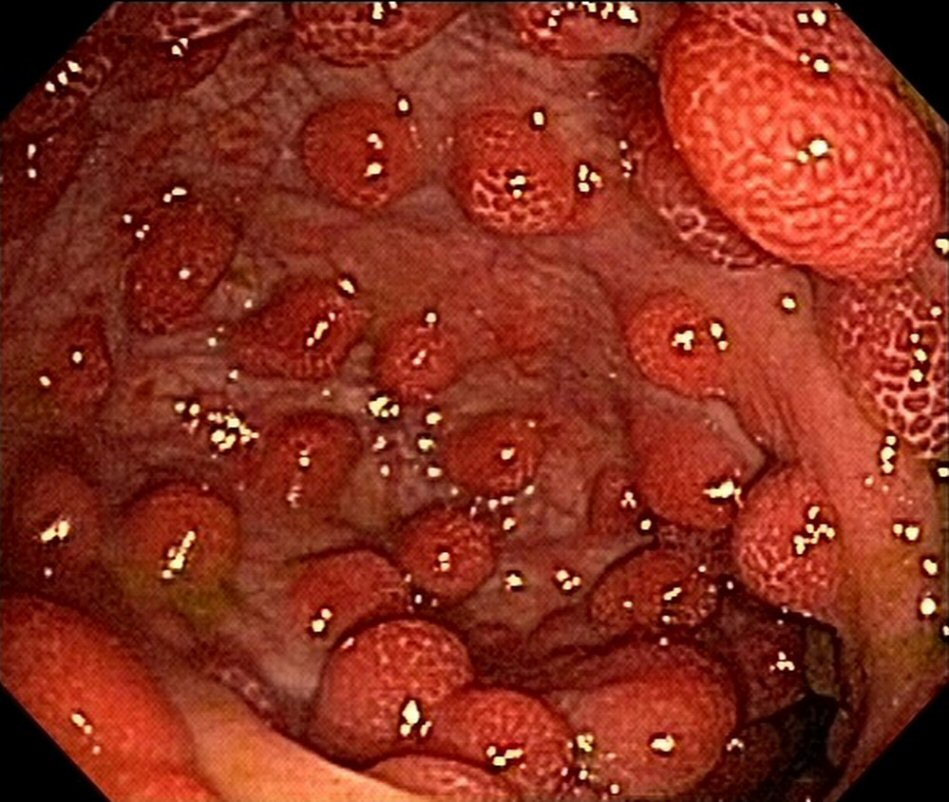 Click for large image | Figure 3. Upper endoscopy reveals polyposis in duodenum at 8-month follow-up. |
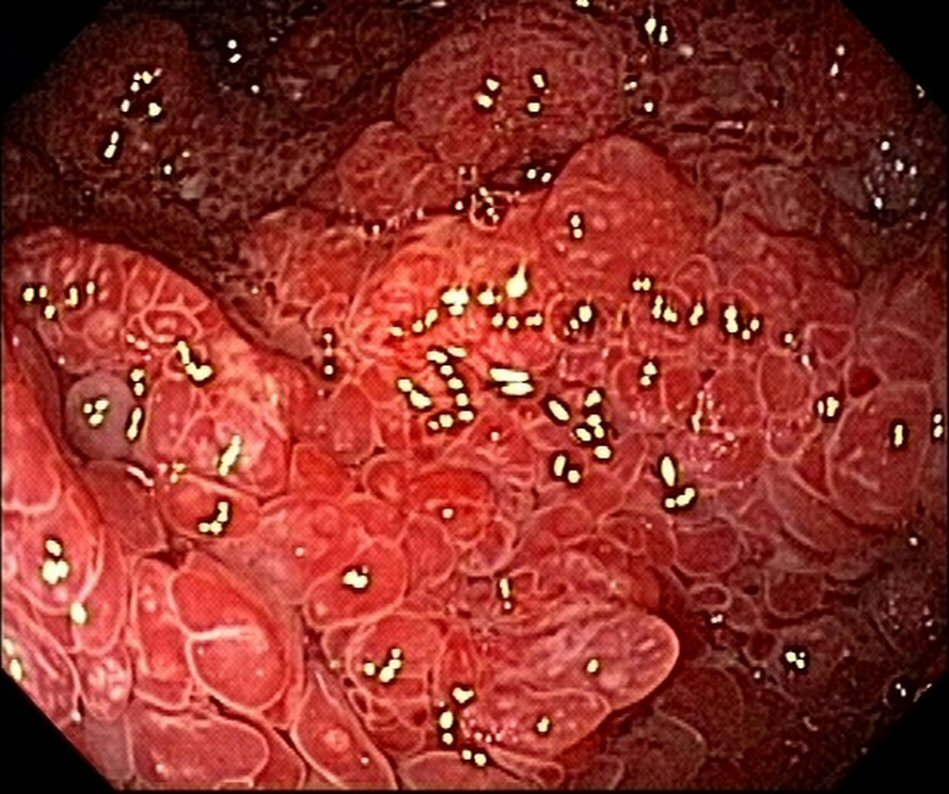 Click for large image | Figure 4. Colonoscopy reveals polyposis in colon and rectum at 8-month follow-up. |
| Discussion | ▴Top |
CCS is a rare nonhereditary condition characterized by nonhereditary gastrointestinal polyposis and skin changes including hyperpigmentation, alopecia and nail disorders [2]. The mean age of presentation is 59 years, but most patients are over 50 years old when the syndrome is identified [3], and may vary from 31 to 85 years [6]. The pathogenesis is unknown [3]. There is confirmation that emotional and physical stresses are important risk factors for this syndrome [7].
Cutaneous manifestations have been attributed to malabsorption and malnutrition caused by gastrointestinal pathology [4]. Several types of diseases concomitant with CCS have also been reported [1]. No mutations of the PTEN tumor suppressor gene, which is located at 10q23.3 and responsible for another gastrointestinal polyposis syndrome, Cowden’s disease, have been in patients with CCS [8].
Most of the patients present all the main manifestations of the syndrome, which are the presence of polyps and the cutaneous triad previously mentioned. The most common initial symptoms are diarrhea and dysgeusia [7]. Diarrhea occurs in large fecal volumes, which may be associated with steatorrhea and melena, usually diarrhea is followed by skin and nail changes. Patients also have total or partial loss of appetite, weight loss, constant or episodic abdominal pain, and weakness. Malabsorption of the gastrointestinal tract leads to malnutrition, anemia, hydroelectrolytic disorders and protein-losing enteropathy [8]. Polyps generally involve the entire gastrointestinal tract, with the exception of the esophagus [3] and are non-neoplastic hamartomas [7].
The clinical course is characterized as a progressive disease with occasional spontaneous remissions and frequent recurrence [4]. Despite the rare correlation of the syndrome with malignant neoplasias [5], there is concern that these polyps may have malignant potential, as evidenced by dysplastic bowel disorders and gastric, colon and rectum cancers in patients with the syndrome [7]. It has been observed that gastric cancer occurred in 10.0% of the cases registered in Japan and that patients with the syndrome have an increased prevalence of gastric cancer in relation to the Japanese general population [8]. The risk of colon cancer may be high because of the difficulties of detecting pre-malignant adenomas because of multiple inflammatory pseudopolypes [4]. Approximately 9-15.0% of the cases were attributed to malignant conditions [5].
It is believed that the Cronkhite-Canada infant syndrome is a variant of juvenile gastrointestinal polyposis, its mode of inheritance is assumed to be autosomal recessive, but the inbreeding of the parents has not been described in any case and may present as sporadic condition. Macrocephaly is a typical sign of infantile syndrome; in addition they develop sympathetic desquamative eruption in the lumbar region, buttocks, genital area, lips and perioral region, similar to cutaneous lesions in acrodermatitis enteropathica [3].
The diagnosis of the syndrome is based on malabsorption characteristics established by characteristic clinical, endoscopic, radiological and histological findings. It is sometimes difficult to diagnose, since polyps of the syndrome and juvenile polyps overlap and may appear histologically identical. The difference between these two types is the mucosa, the polyps of the syndrome reveal abnormalities, such as the presence of cystic glands, edema, congestion and inflammation of lamina own, juvenile polyps are normal. Although the syndrome has characteristic features, the differential diagnosis includes familial adenomatous polyposis, Peutz-Jeghers syndrome, Cowden’s disease and juvenile polyposis [7]. Therefore, the determination of the syndrome would be according to other criteria and not only based on the histology of the polyps [5].
The disease is usually progressive and may present potentially fatal complications, such as malnutrition, gastrointestinal bleeding and infection, which often occur with a 5-year mortality rate of 50.0% [9], if the disease is not treated or if treatment is inadequate or delayed [4]. However, spontaneous regressions were observed in 5-10.0% of cases of the syndrome, regardless of treatment [8].
The best treatment for CCS is currently unknown due to its rarity [8]. There are no evidence-based therapies and there are no studies of medical or surgical interventions to guide the therapy [7]. Current treatments used alone or in combination include steroids, nutritional therapy, acid-5-aminosalicylate, histamine H2-receptor antagonist, anti-tumor necrosis factor (TNF-a), immunomodulators, and Helicobacter pylori eradication. Steroids are considered the main support of medical treatment, although the recommended dose and the duration of its use vary widely in the literature, with no current gold standard [4]. However, the prognosis after treatment with steroids is unknown [9]. The total treatment period is also unknown; recommendations range from 6 to 12 months of combined therapy [8].
Conclusions
We report a case of adolescent CCS that was treated with a combination of steroids and metronidazole. The patient was asymptomatic but with persistent endoscopic abnormalities 8 months after the diagnosis.
Acknowledgments
We greatly appreciate all the support of the Kaiser Clinic of Sao Jose do Rio Preto SP Brazil.
Disclosure
The authors of this case report have no financial interest to disclose.
| References | ▴Top |
- Yuan B, Jin X, Zhu R, Zhang X, Liu J, Wan H, Lu H, et al. Cronkhite-Canada syndrome associated with rib fractures: a case report. BMC Gastroenterol. 2010;10:121.
- Iqbal U, Chaudhary A, Karim MA, Anwar H, Merrell N. Cronkhite-canada syndrome: a rare cause of chronic diarrhea. Gastroenterology Res. 2017;10(3):196-198.
doi pubmed - Parikh K, et al. Cronkhite-Canada Syndrome. Medscape, 2016. Disponivel em: http://emedicine.medscape.com/article/1096789-overview#a4. Acesso 15 de fevereiro de 2017.
- Watanabe C, Komoto S, Tomita K, Hokari R, Tanaka M, Hirata I, Hibi T, et al. Endoscopic and clinical evaluation of treatment and prognosis of Cronkhite-Canada syndrome: a Japanese nationwide survey. J Gastroenterol. 2016;51(4):327-336.
doi pubmed - Safari MT, Shahrokh S, Ebadi S, Sadeghi A. Cronkhite- Canada syndrome; a case report and review of the literature. Gastroenterol Hepatol Bed Bench. 2016;9(1):58-63.
pubmed - Orphanet, Cronkhite-Canada Syndrome, 2009. Disponível em: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=2930&lng=P. Acesso 15 de fevereiro de 2017.
- Sweetser S, Boardman LA. Cronkhite-Canada syndrome: an acquired condition of gastrointestinal polyposis and dermatologic abnormalities. Gastroenterol Hepatol (N Y). 2012;8(3):201-203.
- Kopacova M, Urban O, Cyrany J, Laco J, Bures J, Rejchrt S, Bartova J, et al. Cronkhite-Canada syndrome: review of the literature. Gastroenterol Res Pract. 2013;2013:856873.
doi pubmed - Yamakawa K, Yoshino T, Watanabe K, Kawano K, Kurita A, Matsuzaki N, Yuba Y, et al. Effectiveness of cyclosporine as a treatment for steroid-resistant Cronkhite-Canada syndrome; two case reports. BMC Gastroenterol. 2016;16(1):123.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.