










| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website http://www.gastrores.org |
Original Article
Volume 10, Number 4, August 2017, pages 224-234
Gastrin-Releasing Peptide and Glucose Metabolism Following Pancreatitis
Sayali A. Pendharkara, Marie Drurya, Monika Waliaa, Murray Korcb, Maxim S. Petrova, c
aDepartment of Surgery, University of Auckland, Auckland, New Zealand
bDepartment of Medicine, Biochemistry and Molecular Biology, Division of Endocrinology, Indiana University School of Medicine, the Melvin and Bren Simon Cancer Center and the Pancreatic Cancer Signature Centre, Indianapolis, IN, USA
cCorresponding Author: Max Petrov, Department of Surgery, University of Auckland, Rm 12.085A, Level 12, Auckland City Hospital, Private Bag 92019, Victoria Street West, Auckland 1142, New Zealand
Manuscript submitted July 24, 2017, accepted August 10, 2017
Short title: GRP After an Episode of Pancreatitis
doi: https://doi.org/10.14740/gr890w
| Abstract | ▴Top |
Background: Gastrin-releasing peptide (GRP) is a pluripotent peptide that has been implicated in both gastrointestinal inflammatory states and classical chronic metabolic diseases such as diabetes. Abnormal glucose metabolism (AGM) after pancreatitis, an exemplar inflammatory disease involving the gastrointestinal tract, is associated with persistent low-grade inflammation and altered secretion of pancreatic and gut hormones as well as cytokines. While GRP is involved in secretion of many of them, it is not known whether GRP has a role in AGM. Therefore, we aimed to investigate the association between GRP and AGM following pancreatitis.
Methods: Fasting blood samples were collected to measure GRP, blood glucose, insulin, amylin, glucagon, pancreatic polypeptide (PP), somatostatin, cholecystokinin, gastric-inhibitory peptide (GIP), gastrin, ghrelin, glicentin, glucagon-like peptide-1 and 2, oxyntomodulin, peptide YY (PYY), secretin, vasoactive intestinal peptide, tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein (MCP)-1, and interleukin-6. Modified Poisson regression analysis and linear regression analyses were conducted. Four statistical models were used to adjust for demographic, metabolic, and pancreatitis-related risk factors.
Results: A total of 83 individuals after an episode of pancreatitis were recruited. GRP was significantly associated with AGM, consistently in all four models (P -trend < 0.05), and fasting blood glucose contributed 17% to the variance of GRP. Further, GRP was significantly associated with glucagon (P < 0.003), MCP-1 (P < 0.025), and TNF-α (P < 0.025) - consistently in all four models. GRP was also significantly associated with PP and PYY in three models (P < 0.030 for both), and with GIP and glicentin in one model (P = 0.001 and 0.024, respectively). Associations between GRP and other pancreatic and gut hormones were not significant.
Conclusion: GRP is significantly increased in patients with AGM after pancreatitis and is associated with increased levels of pro-inflammatory cytokines, as well as certain pancreatic and gut hormones. Detailed mechanistic studies are now warranted to investigate the exact role of GRP in derangements of glucose homeostasis following pancreatitis.
Keywords: Gastrin-releasing peptide; Cytokines; Gut hormones; Pancreatic hormones; Post-pancreatitis diabetes mellitus
| Introduction | ▴Top |
Gastrin-releasing peptide (GRP) is a 27 amino acid peptide cleaved, along with neuromedin B and neuromedin C (which are related isoforms), from the 125 amino acid proGRP precursor [1]. This family of peptides, with a highly conserved sequence between species [2], binds to G-protein-coupled receptors with seven transmembrane domains. In humans, there are three receptor subtypes, two of which bind GRP with high affinity - GRP-receptor (GRP-R) and neuromedin-receptor [1]. The third receptor is an orphan bombesin receptor subtype-3 (BRS-3) [3]. GRP binds preferentially to GRP-R, located predominantly in the pancreas, gastrointestinal tract, and central nervous system [2, 4], to play a role in numerous functions [4-7]. For example, GRP acts as a proangiogenic factor and mitogen in certain cancers (in particular, small-cell lung cancer) [8]. It has also been reported to improve intestinal barrier function and oxidative stress, mitigate deleterious effects of sepsis, and decrease inflammation [2, 4]. Increased number of GRP-R has been observed in gastrointestinal inflammatory diseases such as chronic pancreatitis (in autopsied exocrine pancreatic parenchyma) and inflammatory bowel disease (in excised ileal tissue) [9-11]. GRP also has a stimulatory effect on secretion of pancreatic and gut hormones. Intravenous infusion of GRP in fasting state in healthy individuals results in dose-dependent increases in secretion of pancreatic polypeptide (PP), insulin, glucagon, gastrin, gastric-inhibitory peptide (GIP), and cholecystokinin [6, 7, 12]. Further, emerging pre-clinical and clinical evidence also shows that GRP is involved in regulation of blood glucose homeostasis in diabetes mellitus (DM) [1, 5].
DM is a heterogeneous disease encompassing several disorders characterized by persistent hyperglycemia. Diabetes of the exocrine pancreas is a recognized clinical condition [13, 14] that may contribute to up to 10% of all diabetes in the Western population [15, 16] - a statistic based solely on new onset diabetes after chronic pancreatitis and pancreatic cancer. Yet, acute pancreatitis is the most common gastrointestinal disease resulting in hospital admission [17-19], and has the highest incidence rate of 33.74 cases (23.33 - 48.81) per 100,000 person years worldwide compared to other pancreatic diseases [20]. There is an increasing body of evidence that nearly 40% of acute pancreatitis patients develop new onset diabetes after pancreatitis (NODAP) or new onset pre-diabetes [21] and that patients with one attack of acute pancreatitis are at a more than two times greater risk of developing NODAP than the general population [22]. Furthermore, a recent population-based study showed that acute pancreatitis is the most common cause of diabetes of the exocrine pancreas with a prevalence of 78 per 1,000 patients [23]. It is also worth noting that patients with abnormal glucose metabolism (AGM) after pancreatitis have altered secretion of certain pancreatic hormones (insulin, PP, and amylin), gut hormones (oxyntomodulin, glicentin, and vasoactive intestinal peptide (VIP)), and pro-inflammatory cytokines (interleukin-6 (IL-6), monocyte chemoattractant proten-1 (MCP-1), and tumor necrosis factor-α (TNF-α)) [24-26]. While GRP may influence the secretion of pancreatic hormones, gut hormones, and cytokines, the evidence comes largely from pre-clinical studies and dates back to the 1980s [6, 7]. Whether this holds true for patients with AGM after pancreatitis in general and acute pancreatitis in particular, and whether GRP can be used as a marker of NODAP remains to be elucidated.
The primary aim of this study was to investigate the association between GRP and AGM and the effect of potential confounders. The secondary aims were to study the associations between GRP and pancreatic hormones, gut hormones, and pro-inflammatory cytokines.
| Materials and Methods | ▴Top |
Study design
The study was a cross-sectional study of patients after acute pancreatitis. The Health and Disability Ethics Committee (13/STH/182) approved the study protocol.
Study population
Individuals were eligible for the study if they had 1) a primary diagnosis of acute pancreatitis based on two of the following three criteria: abdominal pain suggestive of acute pancreatitis and/or; characteristic radiological finding of acute pancreatitis on computerized tomography or ultrasound scan and/or; serum amylase and/or pancreatic amylase and/or lipase at least three times the upper limit of normal (amylase ≥ 405 U/L; pancreatic amylase ≥ 159 U/L; lipase ≥ 201 U/L) [27]; 2) were at least 18 years of age; 3) resided in Auckland at time of the study; and 4) provided informed consent. All eligible individuals were telephoned and invited to participate in the study. A certified phlebotomist conducted home visits for those participants unable to visit the hospital.
Individuals were not eligible for the study if they had/were pregnant, malignancy, post-endoscopic retrograde cholangiopancreatography pancreatitis, chronic pancreatitis, intraoperative diagnosis of pancreatitis, or pre-diabetes or diabetes before the first hospital admission due to acute pancreatitis.
Definitions
Normal glucose metabolism (NGM) was defined as glycated hemoglobin A1c (HbA1c) ≤ 38 mmol/mol and/or fasting blood glucose (FBG) ≤ 5.5 mmol/L [13].
AGM was defined as newly developed pre-diabetes (HbA1c and/or FBG 39 - 47 mmol/mol and/or 5.6 - 6.9 mmol/L, respectively) or diabetes (HbA1c and/or FBG ≥ 48 mmol/mol and/or ≥ 7.0 mmol/L, respectively) after acute pancreatitis [13].
Body mass index (kg/m2) was determined using a medical scale with stadiometer. Participants were asked to remove any head attire and their shoes for height measurement, and to remove their shoes, jacket, belt, watch, and to empty their pockets of all items for weight measurement.
Etiology was classified, based on participant’s discharge notes, into the following three categories: 1) biliary; 2) alcohol-induced; and 3) other (including but not limited to hypertriglyceridemia, idiopathic pancreatitis, and pancreas divisum).
Severity of acute pancreatitis was determined in line with the 2012 Determinant-Based Classification [28].
Duration was defined as time (in months) from first attack of acute pancreatitis to the time of study.
Recurrence: participants admitted with more than one episode of confirmed acute pancreatitis at the time of the study deemed to have recurrent acute pancreatitis.
Sample acquisition and storage
All study participants were required to visit the clinic at 8:00 am in a fasted state (at least 8 h). Participants were then taken to LabPlus, an International Accreditation New Zealand accredited medical laboratory at Auckland City Hospital where a certified phlebotomist collected venous blood. Where required, appropriate preservatives/inhibitors (mentioned below) were added to the blood samples collected before allowing the blood to clot for at least 30 min before centrifugation. Blood was then centrifuged at 4 °C for 7.5 min at 4,000 g and plasma was separated and stored at -80 °C until use.
Laboratory assays
HbA1c, FBG, and insulin were analyzed at LabPlus, Auckland City Hospital using the boronate affinity chromatography assay (Trinity Biotech, Ireland), enzymatic colourimetric assay (F. Hoffmann-La Roche Ltd), and chemiluminescence sandwich immunoassay (Roche Diagnostics NZ Ltd), respectively.
GRP, gastrin, somatostatin, cholecystokinin, GIP, glicentin, glucagon-like peptide (GLP)-1, GLP-2, oxyntomodulin, VIP, and secretin were measured using the Merck-Millipore ELISA kits in accordance with the user’s manuals. Aprotinin was added into the initial collection step of all aforementioned hormones except GLP-1, to which the DPP-4 inhibitor was added. The Rayto Microplate Reader (V-2100 C, Santa Fe), with an absorbance range of 405 - 630 nm, was used. All results, except for glicentin, were reported in ng/mL. Glicentin was reported in pmol/L.
Glucagon (aprotinin), amylin (protease inhibitor cocktail), ghrelin (serine protease inhibitor), PP, peptide YY (PYY), IL-6, MCP-1, and TNF-α were measured using the MILLIPLEX MAP Human metabolic hormone magnetic bead panel based on the Luminex xMAP technology. The Luminex xPONENT software quantified results (ng/mL) based on the recorded fluorescent reporter signals.
The intra- and inter-assay variation was < 10% and 15%, respectively.
Statistical analyses
All analyses were conducted using SPSS for Windows Version 23 and P-values < 0.05 were considered statistically significant.
The Chi-square test or student's t-test was used to investigate the differences in baseline characteristics between patients with NGM and AGM. Data were presented as frequency or mean ± standard deviation (SD), respectively.
A modified Poisson regression analysis was conducted to investigate the association between GRP and AGM. GRP was categorized into quartiles (quartile 1: 0 - 7.23 ng/mL; quartile 2: 7.24 - 7.91 ng/mL; quartile 3: 7.92 - 8.21 ng/mL; and quartile 4: ≥ 8.22 ng/mL) calculated using the frequency function. The P-trend was calculated by assigning each participant the median value in their quartile and assessing this as a continuous variable. GRP was investigated as an independent variable in one unadjusted and three adjusted models [29]. Model 1 was the unadjusted model and investigated the association between GRP and AGM. Model 2 was adjusted for demographics (age, sex, and ethnicity) and metabolic factors (BMI). Model 3 was adjusted for variables included in model 2 as well as pancreatitis-related factors (etiology, severity of acute pancreatitis, duration since first attack of acute pancreatitis, and recurrence). Model 4 was adjusted for those variables found to be statistically significant in model 3. For these analyses, Pearson’s Chi-square was fit as the scale parameter, accounting for any overdispersion. The offset value was set as 1. For covariance matrix, a robust estimator was selected to derive the most robust estimates. In order to obtain the most conservative estimates, a main-effects model was fit for the one unadjusted and three adjusted models. Data were presented as prevalence ratios (PRs) with corresponding 95% confidence intervals (CIs) and P-values.
Further, having met all assumptions, linear regression analysis was conducted to investigate the association between each pancreatic hormone, gut hormone, and cytokine and GRP. Each pancreatic hormone, gut hormone, and cytokine was investigated as an independent variable in one unadjusted and three adjusted models (as above). Data were presented as β coefficients with corresponding 95% CI and P-values.
| Results | ▴Top |
Study population
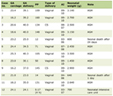
Eighty-three individuals were recruited into the study. Men constituted 60% of the entire cohort. The average age of the study cohort was 51 ± 15 years. Of them, 30 (36%) developed AGM after pancreatitis, while 53 were normoglycaemic. None of the patients that developed AGM had family history of diabetes or received glucose-lowering medications. On average, the study participants were followed up after 30 months since their first attack of acute pancreatitis (Table 1).
 Click to view | Table 1. Baseline Characteristics of Study Participants |
The overall mean ± SD concentrations of each studied pancreatic hormone, gut hormone, and pro-inflammatory cytokine were as follows: amylin, 14.32 ± 27.30; glucagon, 54.42 ± 50.26; insulin, 70.43 ± 48.42; PP, 80.12 ± 103.77; somatostatin, 0.54 ± 0.52; cholecystokinin, 0.92 ± 0.39, GIP, 74.60 ± 87.76; gastrin, 15,572.66 ± 21,845.67; ghrelin, 11.36 ± 10.41; glicentin, 12.64 ± 18.20; GLP-1, 6.76 ± 17.41; GLP-2, 7.01 ± 5.44; oxyntomodulin, 15.06 ± 13.83; P YY, 99.61 ± 120.85; secretin, 0.15 ± 0.05; VIP, 0.52 ± 0.24; IL-6, 9.96 ± 23.73; MCP-1, 93.83 ± 72.37; and TNF-α, 5.15 ± 4.58.
Associations between GRP and glucose metabolism
GRP was significantly associated with AGM in all four models (Fig. 1). Compared with the lowest quartile, the PR (95% CI) of 4.31 (1.34, 13.84; P-trend = 0.017) in the highest quartile differed most in model 4, followed by a PR of 3.70 (1.09, 12.59; P-trend = 0.028) in model 2, a PR of 3.54 (1.19, 10.59; P-trend = 0.042) in model 1, and a PR of 3.35 (1.04, 10.82; P-trend = 0.044) in model 3.
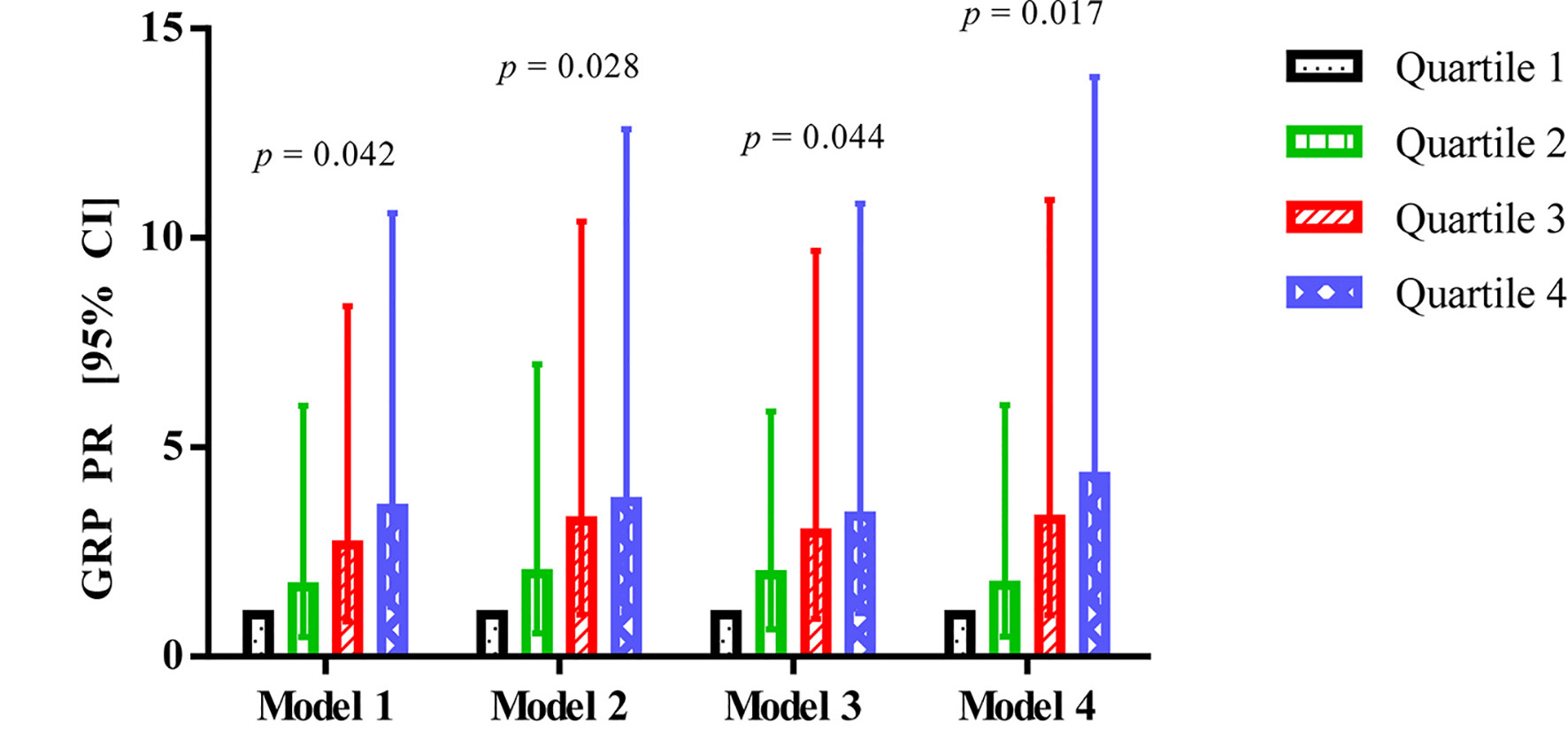 Click for large image | Figure 1. Association between GRP and abnormal glucose metabolism in patients after acute pancreatitis. GRP: gastrin-releasing peptide; CI: confidence interval; PR: prevalence ratio. |
FBG and HbA1c independently contributed to 17.6% (Fig. 2a) and 2.5% (Fig. 2b) of circulating GRP variance, respectively, in the study population.
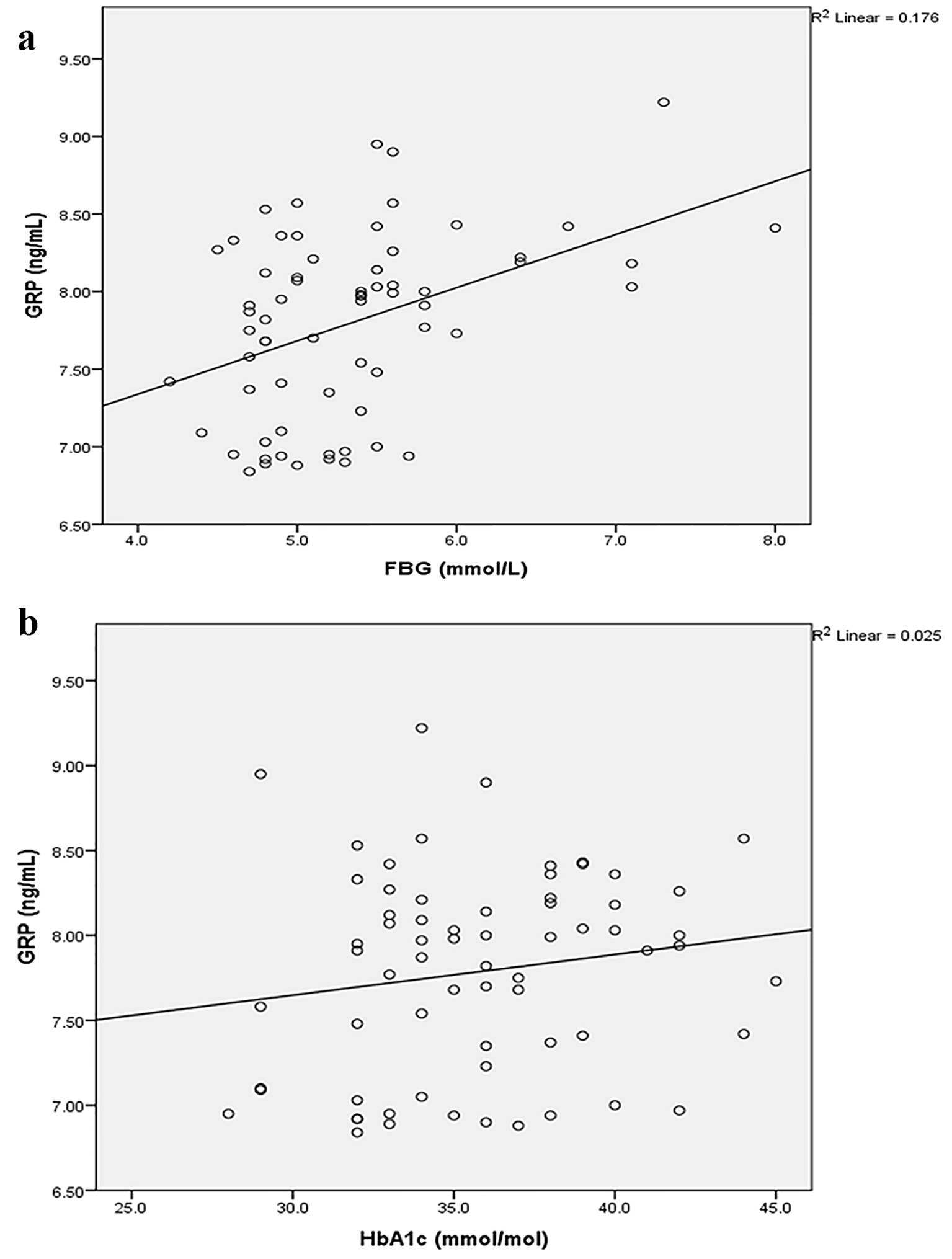 Click for large image | Figure 2. Associations between GRP and FBG (a) and HbA1c (b). FBG: fasting blood glucose; GRP: gastrin-releasing peptide; HbA1c: glycated hemoglobin A1c. |
Associations between GRP and pancreatic hormones
GRP decreased with a change in concentration of glucagon in all four models (Table 2). For every ng/mL change in glucagon, GRP decreased by 0.005 ng/mL in models 3 and 4 (P < 0.001), and by 0.004 ng/mL in models 1 (P = 0.001) and 2 (P = 0.002) (Table 2).
Click to view | Table 2. Associations Between Gastrin-Releasing Peptide and Pancreatic Hormones |
GRP decreased with a change in concentration of PP in the three adjusted models. For every ng/mL change in PP, GRP decreased by 0.002 ng/mL in models 2 (P = 0.010) and 3 (P < 0.001), and by 0.001 ng/mL in model 4 (P = 0.028).
No change in GRP concentration was observed with a change in concentrations of amylin, insulin, and somatostatin in any of the four models (Table 2).
Associations between GRP and gut hormones
GRP decreased with a change in concentrations of PYY, GIP, and glicentin in at least one model (Table 3). For every ng/mL change in PYY, GRP decreased by 0.002 ng/mL in models 3 (P < 0.001) and 4 (P = 0.003), and by 0.001 ng/mL in model 2 (P = 0.026).
Click to view | Table 3. Associations Between Gastrin-Releasing Peptide and Gut Hormones |
GRP decreased with a change in concentrations of glicentin and GIP in one adjusted model. For every ng/mL change in glicentin, GRP decreased by 0.007 ng/mL in model 3 (P = 0.024). For every ng/mL change in GIP, GRP decreased by 0.002 ng/mL in model 3 (P = 0.001).
No change in GRP concentration was observed with a change in concentrations of cholecystokinin, gastrin, ghrelin, GLP-1, GLP-2, oxyntomodulin, secretin, and VIP in any of the four models (Table 3).
Associations between GRP and cytokines
GRP decreased with a change in concentrations of MCP-1 and TNF-α in all four models (Table 4). For every ng/mL change in MCP-1, GRP decreased by 0.003 ng/mL in models 2 (P = 0.001) and 3 (P < 0.001), and by 0.002 ng/mL in models 1 (P = 0.017) and 4 (P = 0.024). For every ng/mL change in TNF-α, GRP decreased by 0.050 ng/mL in model 3 (P < 0.001), by 0.039 ng/mL in model 4 (P = 0.008), by 0.036 ng/mL in model 2 (P = 0.015), and by 0.034 ng/mL in model 1 (P = 0.023).
Click to view | Table 4. Associations Between Gastrin-Releasing Peptide and Cytokines |
No change in GRP concentration was observed with a change in concentration of IL-6 in any of the four models (Table 4).
| Discussion | ▴Top |
GRP has been implicated in glucose homeostasis and in chronic inflammatory diseases such as chronic pancreatitis and inflammatory bowel disease in animal and clinical studies. However, this is the first clinical study to investigate GRP as a marker of AGM in patients following an episode of acute pancreatitis. The findings of this study show that GRP is elevated in patients with AGM after acute pancreatitis and FBG contributes to nearly a fifth of the variance in circulating GRP levels. GRP was also found to be significantly associated with glucagon in the unadjusted and three adjusted models, and with PP, PYY, GIP, glicentin, MCP-1, and TNF-α in at least one adjusted model. These findings have important translational implications and may help devise strategies for the timely identification of individuals with NODAP.
The role of GRP in glucose homeostasis is complex given its expression in the pancreas, gastrointestinal tract, adipose tissue, and central nervous system [30, 31]. Pre-clinical and clinical studies over the past three decades have demonstrated that GRP stimulates the secretion of pancreatic hormones and gut hormones. In the pancreas, GRPergic neurons innervate the islet cells and GRP interacts with the receptors present on the basolateral membranes of these cells to release digestive enzymes. Moreover, the endocrine islet cells express GRP-R, and intra-pancreatic release of GRP by GRPergic neurons has the potential to stimulate the release of hormones such as insulin, glucagon, somatostatin, and PP [12]. While several studies have investigated to various extents the role of certain pancreatic and gut hormones in diabetes of the exocrine pancreas, what mediates the changes in these hormones and ultimately results in NODAP has never been investigated. A recent pre-clinical study by Jha et al examined the effects of intracerebroventricularly administered GRP on glucose metabolism [5]. The authors showed that administration of GRP resulted in persistent hyperglycemia and this was attributed largely to enhanced endogenous glucose production - increased glycogenolysis and/or gluconeogenesis [5]. However, findings from other in vivo studies in rats, mice, and humans showed that GRP potentiates insulin secretion [5, 6, 32-34]. Based on this evidence, and the peripheral and central distribution of GRP neurons, Jha et al concluded that “centrally” administered GRP stimulates glucagon release. A clinical study by Wood et al [6], involving six healthy volunteers, investigated the effect of intravenous GRP infusions on fasting and postprandial glucose levels and pancreatic hormones, namely insulin, glucagon, and PP. While GRP was found to have no effect on basal insulin secretion, it was shown to enhance glucose-dependent insulin secretion. Glucagon and PP levels did not change upon GRP infusion [6]. By contrast, a study by Knigge et al [7], also involving six healthy individuals, reported that PP levels increased by two to four times upon GRP infusion in a dose-dependent manner. Both plasma glucagon and insulin levels were also found to be elevated upon GRP infusion while no change was noted in basal glucose levels [7]. The study by Knigge et al also reported elevated gastrin concentration in response to GRP infusion and the authors speculated that GRP stimulates antral G-cells to stimulate gastrin secretion, which in turn acts on pancreatic islet cells to release pancreatic hormones [7]. However, our study found no association between GRP and gastrin, suggesting that GRP independently regulates glucose homeostasis via the GRP-R present on the islet cells than via the gastrin-mediated pathway. Findings from our study show, for the first time, that GRP levels are increased in patients with AGM after acute pancreatitis and that FBG contributes more than 17% to circulating GRP variance. These findings are a step further towards understanding the intricate and complex pathophysiological mechanisms of glucose derangements in diseases of the exocrine pancreas.
While insulin resistance and compensatory hyperinsulinemia are the primary pathogenetic mechanisms of glucose derangements after acute pancreatitis [24, 35-40], recent evidence also suggests that the elevated insulin levels in blood might be attributed to impaired insulin clearance [41]. This is relevant to the findings of the present study, which showed consistently no association between GRP and insulin in any of the four models but did show consistently significant association between GRP and glucagon in all the models. It is well established that glucagon secretion is inversely related to insulin secretion in pancreatic islets, and that they have opposing effect on circulating blood glucose levels [42]. That GRP was not associated with insulin was a novel finding as evidence to date suggests that GRP stimulates glucose-dependent insulin secretion by acting directly on the pancreatic islet cells [30]. This could be due to the possibility that in individuals with AGM after acute pancreatitis, the GRP and glucagon pathway is preserved, while the insulin pathway is compromised. The reasons for this imbalance are not readily evident. It is unlikely that this could be attributed to desensitized GRP-R on the pancreatic islets since this would compromise both insulin and glucagon secretion. Instead, it is possible that the inflammatory cytokines cause some insulin resistance in target tissues with an inadequate beta cell response. In such a setting, persistent alpha cell activity could exacerbate the hyperglycemia. Whether the elevation in blood glucose levels was due to glycogenolysis or gluconeogenesis, however, could not have been investigated in this study as endogenous glucose production was not measured. GRP is also thought to stimulate insulin secretion by modulating PP [30]. PP has previously been investigated in diseases of the exocrine pancreas, with studies involving chronic pancreatitis patients with and without diabetes, as well as patients with AGM after acute pancreatitis, showing decreased meal-induced and/or fasting PP levels [43-45]. It is likely that the GRP and PP pathway is impaired in individuals with AGM associated with diseases of the exocrine pancreas and that not only decreased PP levels but also elevated GRP levels may be hallmarks of NODAP. This hypothesis needs to be investigated in carefully designed mechanistic studies.
To date, the role of gut hormones, such as GLP-1, GIP, and PYY, has been well investigated in the setting of type 2 diabetes but evidence on the role of these hormones in the setting of NODAP is limited [46]. A recently published study showed that incretins were detectable in 90% of patients after pancreatitis in a fasted state [25]. Given that the effect of food intake was ruled out, it was suggested that other triggers, such as neuropeptides, must have influenced the circulating levels of gut hormones. GRP may fit the bill as this study showed for the first time that GRP is significantly associated with PYY and glicentin. Glicentin, produced in the gut L-cells and central nervous system (brainstem), and containing sequences of glucagon, is known to stimulate insulin secretion [47, 48]. However, a recent study showed that glicentin levels are decreased in patients with AGM after acute pancreatitis [25]. It is possible that the decrease in glicentin levels results in compensatory elevated GRP levels or vice versa. Yet, it is difficult to determine the exact role of glicentin and its association with GRP in AGM after acute pancreatitis as its actions appear to be similar to those of glucagon and GLP-1 [46]. P YY, a member of the PP family, was found to be significantly associated with pro-inflammatory cytokines - IL-6, MCP-1, and TNF-α, in patients after acute pancreatitis [26]. In the present study, PYY was also found to be significantly associated with GRP, which in turn was associated with MCP-1 and TNF-α. Furthermore, there is a known bi-directional relationship between the immune system and nervous system, with peptides such as neuropeptide Y and GRP playing a crucial role [26, 49].
This study has several strengths. First, this is the largest clinical study to investigate the role of GRP and its associations with a comprehensive panel of pancreatic hormones, gut hormones, and the major pro-inflammatory cytokines in defective glucose homeostasis associated with diseases of the exocrine pancreas. Second, the study had a relatively homogenous population of patients after acute pancreatitis - an acute inflammatory disease and the largest contributor to deranged glucose metabolism associated with diseases of the exocrine pancreas [23]. Third, multi-level statistical modeling was used and several patient-, metabolism-, and pancreatitis-related characteristics were adjusted for in order to derive the most robust estimates. The study also has several limitations. First, only circulating insulin levels in blood were measured, which may not estimate insulin secretion accurately. Detailed phenotyping studies, such as hyperglycemic-euglycemic clamps, are now warranted to measure insulin secretion precisely. Also, to determine whether or not GRP stimulates insulin secretion, future studies should administer exogenous GRP at varying doses and investigate its effect on insulin level. Second, while our study showed elevated GRP levels in patients with AGM after acute pancreatitis, and that FBG contributed to 17% of variance in GRP levels, it is not known whether FBG stimulates increased GRP secretion or vice versa. Use of high-performance liquid chromatography glucose tracer is warranted in future studies to measure endogenous glucose production upon administration of GRP [50]. Third, we did not use synthetic GRP analogues/ligands to elucidate whether the increased circulating GRP levels were in response to AGM in patients after acute pancreatitis or desensitized GRP-R on the pancreatic islet cells. Last, we did not measure other ligands known to bind to GRP-R (albeit with less affinity), in particular neuromedin B and neuromedin C, which may explain the elevated circulating GRP levels observed in this study [51].
In conclusion, GRP appears to be a misnomer, at least in the setting of pancreatitis. It does not increase the circulating levels of gastrin but rather is a mediator involved in derangements of glucose homeostasis following pancreatitis. The latter is evidenced by increased GRP levels in patients with AGM, its association with certain pancreatic and gut hormones, as well as pro-inflammatory cytokines, all of which have been implicated in NODAP. It is also worth noting that FBG independently contributes to nearly a fifth of circulating GRP levels. Future studies are now warranted to gain further insights into the role of GRP in diseases of the exocrine pancreas.
Acknowledgments
This study was part of the Clinical and epidemiOlogical inveStigations in Metabolism, nutritiOn, and pancreatic diseaseS (COSMOS) program. COSMOS is supported in part by the Health Research Council of New Zealand (grant 15/035 to Dr. Petrov), which played no role in the study design; collection, analysis, or interpretation of data; or writing of the manuscript.
Conflicts of Interest
None.
| References | ▴Top |
- Feng Y, Guan XM, Li J, Metzger JM, Zhu Y, Juhl K, Zhang BB, et al. Bombesin receptor subtype-3 (BRS-3) regulates glucose-stimulated insulin secretion in pancreatic islets across multiple species. Endocrinology. 2011;152(11):4106-4115.
doi pubmed - Ischia J, Patel O, Shulkes A, Baldwin GS. Gastrin-releasing peptide: different forms, different functions. Biofactors. 2009;35(1):69-75.
doi pubmed - Majumdar ID, Weber HC. Biology and pharmacology of bombesin receptor subtype-3. Curr Opin Endocrinol Diabetes Obes. 2012;19(1):3-7.
doi pubmed - Uche-Nwachi E, Mitchell C. Effect of alloxan-diabetes on gastrin-releasing peptide (grp) immunoreactivity in the gastrointestinal tract, of sprague dawley rats and how this may affect some of the diabetic complications. OJBS. 2007;7:3-7.
doi - Jha PK, Foppen E, Challet E, Kalsbeek A. Effects of central gastrin-releasing peptide on glucose metabolism. Brain Res. 2015;1625:135-141.
doi pubmed - Wood SM, Jung RT, Webster JD, Ghatei MA, Adrian TE, Yanaihara N, Yanaihara C, et al. The effect of the mammalian neuropeptide, gastrin-releasing peptide (GRP), on gastrointestinal and pancreatic hormone secretion in man. Clin Sci (Lond). 1983;65(4):365-371.
doi - Knigge U, Holst JJ, Knuhtsen S, Petersen B, Krarup T, Holst-Pedersen J, Christiansen PM. Gastrin-releasing peptide: pharmacokinetics and effects on gastro-entero-pancreatic hormones and gastric secretion in normal men. J Clin Endocrinol Metab. 1984;59(2):310-315.
doi pubmed - Mattei J, Achcar RD, Cano CH, Macedo BR, Meurer L, Batlle BS, Groshong SD, et al. Gastrin-releasing peptide receptor expression in lung cancer. Arch Pathol Lab Med. 2014;138(1):98-104.
doi pubmed - Fleischmann A, Laderach U, Friess H, Buechler MW, Reubi JC. Bombesin receptors in distinct tissue compartments of human pancreatic diseases. Lab Invest. 2000;80(12):1807-1817.
doi pubmed - Petronilho F, Danielski LG, Roesler R, Schwartsmann G, Dal-Pizzol F. Gastrin-releasing peptide as a molecular target for inflammatory diseases: an update. Inflamm Allergy Drug Targets. 2013;12(3):172-177.
doi pubmed - ter Beek WP, Muller ES, Van Hogezand RA, Biemond I, Lamers CB. Gastrin releasing peptide receptor expression is decreased in patients with Crohn's disease but not in ulcerative colitis. J Clin Pathol. 2004;57(10):1047-1051.
doi pubmed - Konturek SJ, Zabielski R, Konturek JW, Czarnecki J. Neuroendocrinology of the pancreas; role of brain-gut axis in pancreatic secretion. Eur J Pharmacol. 2003;481(1):1-14.
doi pubmed - American Diabetes Association. (2) Classification and diagnosis of diabetes. Diabetes Care. 2015;38(Suppl):S8-S16.
doi - Petrov MS. Diabetes of the exocrine pancreas: American Diabetes Association-compliant lexicon. Pancreatology. 2017;17(4):523-526.
doi pubmed - Ewald N, Kaufmann C, Raspe A, Kloer HU, Bretzel RG, Hardt PD. Prevalence of diabetes mellitus secondary to pancreatic diseases (type 3c). Diabetes Metab Res Rev. 2012;28(4):338-342.
doi pubmed - Ewald N, Bretzel RG. Diabetes mellitus secondary to pancreatic diseases (Type 3c)--are we neglecting an important disease? Eur J Intern Med. 2013;24(3):203-206.
doi pubmed - Fagenholz PJ, Castillo CF, Harris NS, Pelletier AJ, Camargo CA Jr. Increasing United States hospital admissions for acute pancreatitis, 1988-2003. Ann Epidemiol. 2007;17(7):491-497.
doi pubmed - Goldacre MJ, Roberts SE. Hospital admission for acute pancreatitis in an English population, 1963-98: database study of incidence and mortality. BMJ. 2004;328(7454):1466-1469.
doi pubmed - O’Farrell A, Allwright S, Toomey D, Bedford D, Conlon K. Hospital admission for acute pancreatitis in the Irish population, 1997 2004: could the increase be due to an increase in alcohol-related pancreatitis? J Public Health (Oxf). 2007;29(4):398-404.
doi pubmed - Xiao AY, Tan ML, Wu LM, Asrani VM, Windsor JA, Yadav D, Petrov MS. Global incidence and mortality of pancreatic diseases: a systematic review, meta-analysis, and meta-regression of population-based cohort studies. Lancet Gastroenterol Hepatol. 2016;1(1):45-55.
doi - Das SL, Singh PP, Phillips AR, Murphy R, Windsor JA, Petrov MS. Newly diagnosed diabetes mellitus after acute pancreatitis: a systematic review and meta-analysis. Gut. 2014;63(5):818-831.
doi pubmed - Shen HN, Yang CC, Chang YH, Lu CL, Li CY. Risk of diabetes mellitus after first-attack acute pancreatitis: a national population-based study. Am J Gastroenterol. 2015;110(12):1698-1706.
doi pubmed - Pendharkar SA, Mathew J, Petrov MS. Age- and sex-specific prevalence of diabetes associated with diseases of the exocrine pancreas: A population-based study. Dig Liver Dis. 2017;49(5):540-544.
doi pubmed - Pendharkar SA, Asrani VM, Xiao AY, Yoon HD, Murphy R, Windsor JA, Petrov MS. Relationship between pancreatic hormones and glucose metabolism: A cross-sectional study in patients after acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2016;311(1):G50-58.
doi pubmed - Pendharkar SA, Asrani VM, Murphy R, Cutfield R, Windsor JA, Petrov MS. The role of gut-brain axis in regulating glucose metabolism after acute pancreatitis. Clin Transl Gastroenterol. 2017;8(1):e210.
doi pubmed - Pendharkar SA, Singh RG, Petrov MS. Cross-talk between innate cytokines and the pancreatic polypeptide family in acute pancreatitis. Cytokine. 2017;90:161-168.
doi pubmed - Petrov MS. Editorial: abdominal fat: a key player in metabolic acute pancreatitis. Am J Gastroenterol. 2013;108(1):140-142.
doi pubmed - Dellinger EP, Forsmark CE, Layer P, Levy P, Maravi-Poma E, Petrov MS, Shimosegawa T, et al. Determinant-based classification of acute pancreatitis severity: an international multidisciplinary consultation. Ann Surg. 2012;256(6):875-880.
doi pubmed - Gelman A, Hill J, Yajima M. Why we (usually) don't have to worry about multiple comparisons. J Res Educ Eff. 2012;5:189-211.
doi - Paula GS, Souza LL, Bressane NO, Maravalhas R, Wilieman M, Bento-Bernardes T, Silva KR, et al. Mice with deletion of neuromedin b receptor exhibit decreased oral glucose-stimulated insulin release. Horm Metab Res. 2016;48(12):854-861.
doi pubmed - Monstein HJ, Grahn N, Truedsson M, Ohlsson B. Progastrin-releasing peptide and gastrin-releasing peptide receptor mRNA expression in non-tumor tissues of the human gastrointestinal tract. World J Gastroenterol. 2006;12(16):2574-2578.
doi pubmed - Hermansen K, Ahren B. Gastrin releasing peptide stimulates the secretion of insulin, but not that of glucagon or somatostatin, from the isolated perfused dog pancreas. Acta Physiol Scand. 1990;138(2):175-179.
doi pubmed - Pettersson M, Ahren B. Gastrin releasing peptide (GRP): effects on basal and stimulated insulin and glucagon secretion in the mouse. Peptides. 1987;8(1):55-60.
doi - Pettersson M, Ahren B. Insulin and glucagon secretion in the rat: effects of gastrin releasing peptide. Neuropeptides. 1988;12(3):159-163.
doi - Andersson B, Pendse ML, Andersson R. Pancreatic function, quality of life and costs at long-term follow-up after acute pancreatitis. World J Gastroenterol. 2010;16(39):4944-4951.
doi pubmed - Balzano G, Dugnani E, Pasquale V, Capretti G, Radaelli MG, Garito T, Stratta G, et al. Clinical signature and pathogenetic factors of diabetes associated with pancreas disease (T3cDM): a prospective observational study in surgical patients. Acta Diabetol. 2014;51(5):801-811.
doi pubmed - Das SL, Kennedy JI, Murphy R, Phillips AR, Windsor JA, Petrov MS. Relationship between the exocrine and endocrine pancreas after acute pancreatitis. World J Gastroenterol. 2014;20(45):17196-17205.
doi pubmed - Kennedy JI, Askelund KJ, Premkumar R, Phillips AR, Murphy R, Windsor JA, Petrov MS. Leptin is associated with persistence of hyperglycemia in acute pancreatitis: a prospective clinical study. Medicine (Baltimore). 2016;95(6):e2382.
doi pubmed - Wu D, Xu Y, Zeng Y, Wang X. Endocrine pancreatic function changes after acute pancreatitis. Pancreas. 2011;40(7):1006-1011.
doi pubmed - Yki-Jarvinen H, Kiviluoto T, Taskinen MR. Insulin resistance is a prominent feature of patients with pancreatogenic diabetes. Metabolism. 1986;35(8):718-727.
doi - Zalewski L. [Long-term follow up of acute and chronic pancreatitis during insulin and peptide C secretion and carbohydrate tolerance]. Pol Arch Med Wewn. 1996;96(6):561-569.
pubmed - Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A. 2010;107(37):16009-16012.
doi pubmed - Andersen BN, Hagen C, Klein HC, Stadil F, Worning H. Correlation between exocrine pancreatic secretion and serum concentration of human pancreatic polypeptide in chronic pancreatitis. Scand J Gastroenterol. 1980;15(6):699-704.
doi pubmed - Mee AS, Klaff LJ, Girdwood AH, Paul M, Tyler M, Marks IN. Comparative study of pancreatic polypeptide (PP) secretion, endocrine and exocrine function, and structural damage in chronic alcohol induced pancreatitis (CAIP). Gut. 1983;24(7):642-647.
doi pubmed - Rickels MR, Bellin M, Toledo FG, Robertson RP, Andersen DK, Chari ST, Brand R, et al. Detection, evaluation and treatment of diabetes mellitus in chronic pancreatitis: recommendations from PancreasFest 2012. Pancreatology. 2013;13(4):336-342.
doi pubmed - Sandoval DA, D'Alessio DA. Physiology of proglucagon peptides: role of glucagon and GLP-1 in health and disease. Physiol Rev. 2015;95(2):513-548.
doi pubmed - Panaro BL, Tough IR, Engelstoft MS, Matthews RT, Digby GJ, Moller CL, Svendsen B, et al. The melanocortin-4 receptor is expressed in enteroendocrine L cells and regulates the release of peptide YY and glucagon-like peptide 1 in vivo. Cell Metab. 2014;20(6):1018-1029.
doi pubmed - Latorre R, Sternini C, De Giorgio R, Greenwood-Van Meerveld B. Enteroendocrine cells: a review of their role in brain-gut communication. Neurogastroenterol Motil. 2016;28(5):620-630.
doi pubmed - Hernanz A, Tato E, De la Fuente M, de Miguel E, Arnalich F. Differential effects of gastrin-releasing peptide, neuropeptide Y, somatostatin and vasoactive intestinal peptide on interleukin-1 beta, interleukin-6 and tumor necrosis factor-alpha production by whole blood cells from healthy young and old subjects. J Neuroimmunol. 1996;71(1-2):25-30.
doi - Bavenholm PN, Pigon J, Ostenson CG, Efendic S. Insulin sensitivity of suppression of endogenous glucose production is the single most important determinant of glucose tolerance. Diabetes. 2001;50(6):1449-1454.
doi pubmed - Roesler R, Schwartsmann G. Gastrin-releasing peptide receptors in the central nervous system: role in brain function and as a drug target. Front Endocrinol (Lausanne). 2012;3:159.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.